Chapter 56 The brain and cerebral visual impairment
Developmental defects
Embryological errors
Cephalocele (encephalocele)
1. Occipital cephaloceles. In these portions of the occipital cortex and the occipital horn of the lateral ventricle herniate into the defect. Severe visual defects are associated both with the anomaly and the results of any surgical correction.
2. Frontal ethmoidalcephaloceles. These do not usually impinge on any visual structures, but ipsilateral optic nerve dysplasia may be associated with them.
3. Nasal pharyngeal cephaloceles. These are uncommon, but visual function is almost always affected. The optic nerves and chiasm may be compromised as they are stretched when they extend into the sac of the defect. Optic nerve hypoplasia and retinal dysplasia and coloboma are frequent associations.
Holoprosencephaly
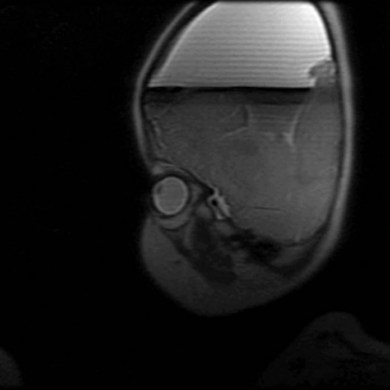
During the second month of gestation the forebrain (prosencephalon) is cleaved transversely into the telencephalon and diencephalon and sagittally into the cerebral hemispheres and lateral ventricles. Failure of differentiation and cleaving of the prosencephalon results in a group of disorders referred to as “the holoprosencephalies.” These are caused by both teratogens and genetic factors. Maternal diabetes is the most common recognized teratogen. Holoprosencephaly can be seen in a number of syndromes including Patau’s syndrome (trisomy 13), Edwards’ syndrome (trisomy 18), and de Morsier’s syndrome. Facial dysmorphism (hypotelorism and midline clefts) and numerous central nervous system anomalies are frequently associated (Fig. 56.1). Corpus callosum dysgenesis is common; unsurprisingly, therefore, so is optic nerve hypoplasia (see Chapter 51). Indeed, a significant subset of patients with septo-optic dysplasia may have a mild form of lobar holoprosencephaly.
Malformations of cortical development
Between the second and fourth gestational months, the neurons in the ventricular and subventricular zones of the lateral ventricles proliferate and migrate to the cortical plates. This area of cell proliferation is known as the germinal matrix; here, stem cells give rise to the neurons and glial cells that will form the mature brain. Neurons in early embryogenesis migrate relatively short distances; neurons later in development migrate long distances across the intermediate zones. Neurons arriving first in the cortical mantle assume the deepest locations while later arriving neurons assume a more superficial location. Migration is facilitated by radial glial cells acting as guidelines. After arriving in the cortex neurons form discrete lamina and begin to establish synaptic connection with local and distant neurons. Aberrations in this normal migration and development process result in important neural abnormalities.1 In general, the resulting malformations can be divided into three categories − those due to:
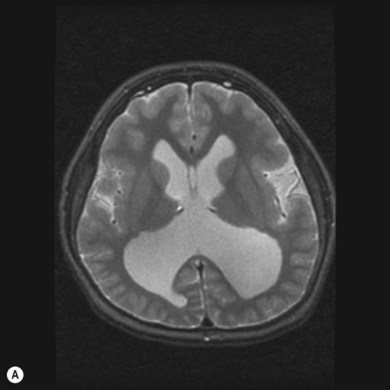
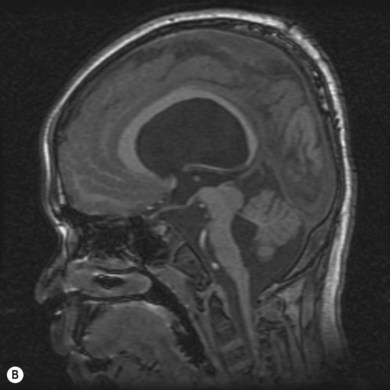
a. Fukuyama-type congenital muscular dystrophy: affected children develop high myopia and a retinal degeneration (Fig. 56.2).
b. The muscle−brain−eye syndrome: this is associated with myopia and early-onset glaucoma and cataracts.
b. The Walker-Warburg syndrome: this comprises microphthalmos, glaucoma, optic nerve anomalies, and retinal dysplasia and non-attachment.
Polymicrogyria is the most common malformation of cortical development. It occurs as the result of the interruption of the late stages of neuron migration during the stages of cortical organization. The neurons reach the cortical mantle, but the deep layers are distributed which results in disorganized, multiple, and small gyri. Its effect on neurological function is less severe than the proliferation and migration anomalies. It may occur as an isolated focal anomaly of little consequence. The most common area is around the Sylvian fissure. However, it may also occur as a diffuse disorder affecting the entire brain. As an isolated and focal disorder it has been associated with congenital hemianopia (occipital cortex) and dyslexia (left frontal and temporal cortex). It is an important feature of several genetic syndromes including Aicardi’s syndrome, Joubert’s syndrome, Zellweger’s spectrum, Sturge-Weber syndrome, X-linked hydrocephalus, and 22q11.2 deletion syndrome. It is also associated with numerous metabolic disorders.2
Schizencephaly (agenetic porencephaly) is characterized by full thickness clefts spanning the wall of the cerebral hemispheres that are lined by gray matter and often surrounded by polymicrogyric cortex. The pathogenesis is incompletely understood although it is thought to represent an in utero injury to the germinal matrix during the second trimester before the hemispheres form. Previous reports that schizencephaly may be associated with mutations of the EMX2 homeobox gene, located on chromosome 10q26, and expressed in the germinal matrix has been challenged.3 The clefts may be unilateral or bilateral; the lips of the clefts may be open or closed. Seizures, hemiplegia, and mental retardation are the most common symptoms. Involvement of the occipital cortex is unusual but, when it occurs, especially when the cleft’s lips are fused, a homonymous hemianopia may occur. In contrast, patients with bilateral clefts are more likely to be severely retarded, severely limited by motor problems and blindness. The blindness is not usually cortical in origin, but due to optic nerve hypoplasia, a frequent accompanying anomaly in all forms of schizencephaly.
Congenital hemianopia
Children with congenital hemianopia show little evidence of visual dysfunction; the visual field defect is often discovered later in life on a routine eye examination. There may be a history of frequent bicycle or auto accidents, but many patients experience little effect on their daily lives. Therefore, the history may not be helpful in suspecting the presence of a congenital hemianopia. However, there are certain associated ophthalmologic and systemic findings that should alert the ophthalmologist to the possibility of a congenital hemifield defect.4
Most children with a congenital, but not acquired, hemianopia, turn their face toward the defective field when fixing on a target straight ahead of them (see Chapter 81). It is not clear how this compensates for the field defect, although the intact field is “centered” on the body by this maneuver. In any case, a persistent face turn in a child without incomitant strabismus or nystagmus with a null zone (the more common ocular causes of such a turn) should prompt an evaluation of the visual fields. In some cases, the face turn is accompanied by a constant, non-alternating exotropia;5 the exotropic eye is ipsilateral to the field defect (see Chapter 78). Theoretically, a large angle exotropia might significantly expand the binocular visual field with the appropriate sensory adaptation (harmonious anomalous retinal correspondence) although the evidence that it does so is incomplete. The frequent occurrence of exodeviations in neurologic disorders raises the question of the specificity of this finding. However, the unique combination of a face turn and exotropia ipsilateral to the face turn is highly suggestive of congenital hemianopia.
In addition to the visual field defect, face turn, and possible exodeviation, the most prominent ophthalmological finding in these children is localized changes in the optic disc and nerve fiber layer − “homonymous hemioptic atrophy” (hypoplasia). There is loss of retinal nerve fibers in the retinal sectors nasal and temporal to the disc in the eye contralateral to the field defect; in the ipsilateral eye the loss is superior and inferior. This subtle pattern of nerve fiber loss can be seen ophthalmoscopically or with OCT (optical coherence tomography).6 Examination of the optic discs will reveal a band-shaped atrophy of the contralateral disc and temporal atrophy of the ipsilateral disc. Since the lesions causing congenital hemianopia are almost always posterior to the lateral geniculate body, this points to transsynaptic degeneration of the retinogeniculate striate pathways. This has been thought to only occur with prenatal or perinatal insults, but recent experiments in non-human primates and OCT studies in patients with acquired hemianopias suggest that this is not the case.7
Isolated congenital hemianopias are typically caused by developmental anomalies of the occipital cortex.8 These include:
• Occipital lobe dysplasia: abnormality of neuronal/glial proliferation
• Occipital lobe pachygyria: abnormality of neuronal migration
• Occipital lobe polymicrogyria: abnormality of cortical organization
• Porencephaly with or without polymicrogyria
• Gangliogliomas: neoplastic abnormality of neuronal/glial proliferation
Patients with congenital hemianopia are less visually disabled than those with acquired hemianopia. Our understanding of their adaptations and compensations accounting for their minimal disability is incomplete. The possibility that a face turn and/or exotropia could be compensatory has been cited above. A unique saccadic strategy limited to patients with congenital hemianopia allows them to search the blind field. This is distinctly more efficient than the multiple hypometric saccades made by the patient with an acquired hemianopia, but it can occur in congenital lesions ( Video 56.1). Moreover, in children with acquired hemianopias the reaction time to initiate a saccade to explore the blind field is prolonged; this is not the case in congenital hemianopia.9
Video 56.1). Moreover, in children with acquired hemianopias the reaction time to initiate a saccade to explore the blind field is prolonged; this is not the case in congenital hemianopia.9
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


