Teratology of the Eye
Y. Robert Barishak
Abraham Spierer
It is difficult to classify the ocular teratologic conditions by chronology as well as by topography; the findings overlap frequently and, in addition, a correspondence between etiology and findings does not always exist.
This chapter is limited to anomalies and deviations from normal. Molecular genetics have shown that variations occur within each species; therefore, we must be able to differentiate between variation and anomaly.
The causes of malformations are multiple and difficult to recognize. However, genetic abnormalities constitute the majority of congenital malformations, and these consist mostly of improper chromosomal deviations. In some cases, environmental factors (teratogenic substances, nutritional disturbances, irradiation, intrauterine infections, hormonal imbalance, advanced age in mothers, etc.) also play an important role.1 Recently, emphasis has been placed more on multifactorial causation, emphasizing the importance of these environmental causes.
We review the ocular anomalies in two sections. In the first section, we review the anomalies according to their etiology: (a) anomalies derived from improper chromosomal deviations, (b) the craniofacial dysostosis anomalies derived from errors in the development of neural crest cells, (c) anomalies encountered in drug embryopathies, and (d) anomalies encountered in infectious embryopathies.
In the second section, we review the anomalies associated with the different developing tissues of the eye. The time at which the teratogenic insult takes place is critical. During the early periods of embryogenesis and organogenesis, insult affects the germ layers; survival of the embryo is unusual. If the teratogenic insult takes place during the process of differentiation, more limited and specific abnormalities occur. It must be emphasized that abnormalities caused by a teratogenic insult differ according to the degree of severity of the insult: the more severe the insult, the more severe the anomaly.
SECTION 1
Anomalies Resulting from Improper Chromosomal Deviations
Anomalies resulting from improper chromosomal deviations present some common systemic entities such as low birth weight, failure to thrive, mental retardation, behavioral disorders, congenital heart disease, teeth and kidney abnormalities, skeletal malformations, hypertelorism, persistence of embryonic and fetal hemoglobin, and abnormal dermatoglyphic findings. Ocular manifestations are prominent.
Trisomies
The occurrence of a supernumerary autosomal chromosome is the most commonly seen anomaly. Trisomies consist of a partial or complete duplication of the chromosomes 2, 4, 13, 17, 18, 21, 22, X, and Y.
Trisomy 13 also known as trisomy D1, Patau syndrome, or Bartholin syndrome, is due to a nondisjunction of the chromosome 13. The main ocular symptoms are cyclopia, microphthalmos, uveal coloboma, persistent hyperplastic primary vitreous, glaucoma and undifferentiation of the angle structures, cataract, corneal opacities, retinal dysplasia, and optic nerve hypoplasia. The major systemic findings include microcephaly, arhinencephaly, ventricular septal defects, unguinal and umbilical hernias, polydactyly, and rudimentary digits in hands and feet.
Trisomy 21 (Down syndrome, mongolism) is a wellknown anomaly induced by a nondisjunction of chromosome 21, sometimes associated with a translocation of its long arm with the long arm of chromosomes 13, 14, 15, or another chromosome. The incidence increases with maternal age. Ocular findings include a mongoloid slant of the eyelids, cataract, high refractive errors, and keratoconus. Systemic findings include the characteristic short stature and short neck, brachycephaly, delayed fontanelle closure, small external ears, tendency to open the mouth, hypotonia, small broad hands and feet, genital anomalies such as undescended testes and enlarged labia, ovarian hypoplasia, cardiac anomalies, and mental retardation.
Trisomy 18, also known as trisomy E or Edwards syndrome is due to a nondisjunction of chromosome 18. Ocular signs include microphthalmos, congenital glaucoma, uveal coloboma, pigment disturbances, optic nerve anomalies, hypertelorism, blepharophimosis, epicanthus, ptosis, and corneal opacities. Systemic findings include the characteristic facies with low-set, malformed ears; micrognathia; microstomia; decreased growth rate; and cardiac anomalies.
Trisomy X (Klinefelter syndrome) is due to a duplication of the X chromosome that causes an incomplete expression of male characteristics, with underdeveloped testes and gynecomastia. Ocular anomalies are rare except for coloboma, which is apparently the result of a teratogenic insult prior to the closure of the embryonic fissure.
Trisomy A is essentially a partial Trisomy 2q. Ocular symptoms include uveal colobomas, congenital glaucoma, lens dislocation, exotropia, and mandibulofacial dysostosis. Systemic findings involve the face, mouth, and digits and include also hypertelorism and microcephaly. This anomaly is compatible with life.
Trisomy of Chromosome 4 in Group B involves the short arm of chromosome 4. Ocular symptoms include uveal colobomas and blepharophimosis. Systemic findings include microcephaly or hydrocephaly and facial anomalies such as low-set ears, micrognathia, and macroglossia.
Duplication of 10q and 10p Chromosome presents with ocular findings such as an antimongoloid slant of the lid fissure, ptosis, epicanthus, hypertelorism, blepharophimosis, and microphthalmos. Systemic findings include a characteristic facies with a broad, flat, nasal bridge, small nose and bow-shaped mouth, flexion deformities in the wrists, abnormal genitalia, low birth weight, and severe mental retardation. Babies with this anomaly usually do not survive.
Trisomy 17 presents with uveal colobomas, an antimongoloid slant of the eyelids, epicanthus, hypertelorism, and anomalies of the optic nerve head. Systemic findings include cardiovascular anomalies, finger anomalies, spasticity, and seizures.
Trisomy 22 (cat’s eye syndrome) can be seen in cases of partial trisomy 13. Ocular anomalies include preauricular fistulae, an antimongoloid slant of the eyelids and low-set ears, hypertelorism, exotropia, myopia, cataract, retinal degeneration, and Sturge-Weber syndrome. Systemic signs include anal atresia.
Anomalies Resulting from Chromosomal Deletions
A chromosomal deletion consists of the dysjunction of a whole or fragment of a chromosome during meiosis. The most common ones are discussed here.1
Turner syndrome is caused by a lack of an X chromosome. In some cases, a short arm deletion of the X chromosome occurs; in others, a partial or complete deletion of all the arms of the X chromosome occurs. This results in the formation of a ring chromosome.2 This deletion causes an incomplete expression of female sex characteristics. Ocular signs include microcornea, eccentric pupil, epicanthus, and cataract. Systemic findings include short stature, webbing of the neck, prominent ears, vascular and skeletal anomalies, micrognathia, and mental retardation.1
Wolf Syndrome involves the deletion of chromosome 4p. The deletion occurs in the small arm of chromosome 4. Ocular signs include an antimongoloid slant of the eyelids, ptosis, uveal coloboma, and defects in the medial half of the brow. Systemic findings include mental retardation, seizures, microcephaly, and hypotonia.
Cri du Chat Syndrome is due to the deletion of the small arm of chromosome 5. This deletion causes the following ocular signs: hypertelorism, epicanthus, lid coloboma, decreased tear formation and occasionally microphthalmos, ocular dermoid, and antimongoloid slant of the eyelids. Among the systemic findings, the most typical is the characteristic vocalization of the patient, which resembles the mewing of a cat. The patient also presents with craniofacial malformations, micrognathia, microcephaly, hypotony, mental retardation, and skeletal anomalies such as short metacarpal bones.
13q Deletion Syndrome is due to the deletion of the long arm of the chromosome 13. Ocular anomalies include microphthalmos, coloboma, hypertelorism, epicanthus, ptosis, and occasionally retinoblastoma. Systemic signs include microcephaly, mental retardation, low-set ears, micrognathia, congenital heart disease, undescended testes, and abnormal digits.
Chromosome 18q Deletion is also known as De Grouchy syndrome. The basic abnormality is a partial deletion of chromosome 18. The deletion might involve the short or long arm, or both the short and long arms of chromosome 18. Ocular anomalies include microphthalmos, microcornea, corneal opacity, uveal coloboma, glaucoma, macular abnormality, nystagmus, hypertelorism, epicanthus, ptosis, and strabismus. Systemic findings include microcephaly, short stature, mental retardation, low-set ears, and deafness.
Chromosome 11 Deletion was described by Yanoff and Fine2 as the AGR triad (aniridia, genitourinary, mental retardation syndrome). Deletion of chromosome 11q results in trigonocephaly, a broad nasal bridge, and micrognathia. However, the best known manifestations are aniridia and Wilms tumor.
Triploidy
Triploidy is the most common chromosomal aberration.1 The patient has 69 chromosomes (instead of the normal 64). It is usually caused by an extra set of chromosomes, possibly because the ovum has been fertilized by two spermatozoid cells. Hypoxia, hormonal therapy before or during pregnancy, advanced maternal age have been suggested as possible causes. Most embryos abort spontaneously. Those who survive present hypertelorism, blepharophimosis, uveal coloboma, microcornea, persistence of hyaloid vessels, retinal dysplasia, and optic atrophy.
Mosaicism
Chromosomal mosaicism implies the presence of two or more populations of karyotypically different chromosomes in cells from a single individual.2 During their development, embryonal cells may present anomalies in their mitotic activity. One is mitotic nondisjunction, in which the replicated chromosome fails to separate, and the other is anaphase lag, in which the replicated and separated chromosome fails to migrate. Mitotic nondisjunction may occur early in gestation, when the population of cells is small, and then the abnormal chromosomes become lost during development. It may occur at an advanced stage of embryonal development when the population of cells is large, but then the number of abnormal chromosomes constitutes a minority in the total cell population; subsequently, they have no effect on the development of the embryo.
However, if such abnormal chromosomes occur in cells of the reproductive organs, some of the gametes may carry abnormal chromosomes and, as a result, mosaic parents might present a higher risk for having chromosomally abnormal children.3
Tetraploid-Diploid Mosaicism has two distinct populations of cells; large cells with 92 chromosomes (tetraploid) and normal-sized cells with 46 chromosomes (diploid). Ocular anomalies include microphthalmos, corneal opacities, and leukocoria. Systemic findings include microcephalus, developmental arrest of the brain, cardiovascular anomalies, and finger and toe deformities. These patients have a short life span.
Craniofacial Dysostosis Anomalies Derived from Errors in Neural Crest Cell Development
The importance of the cephalic neural crest cells in the morphogenesis of the craniofacial and orbital tissues has been stressed by many authors.4, 5, 6 and 7 Couly and colleagues4 proposed an embryologic classification of facial malformations based on embryonic development disorders of neural crest-derived cells of cephalic origin that constitute the craniofacial ectomesenchyme. They refer to these malformations as neurocristopathies. Following Edwards’ review of these malformations, we classify the craniofacial dysplastic syndromes into (a) clefting disorders, (b) ectodermal mesenchymal induction anomaly, (c) craniostenosis, and (d) craniofacial syndromes.5
Clefting Defects
The most prominent midfacial anomaly is the median cleft syndrome, which appears with hypertelorism, median cleft nose, median cleft lip, median cleft premaxilla, and cranium bifidum. This syndrome may be associated with median lipomas, median teratomas, and frontal encephaloceles. The median cleft face syndrome may present four facial patterns, according to the degree of severity; it quite often is associated with a normal ocular structure. Severely affected patients do not survive.
Maldevelopment also may result in lateral, transverse, and oblique facial clefts. These defects also may include the lateral aspect of the mouth and portions of the lower lid and ear. Lid colobomas and cleft palate also may be seen. These defects may be termed oro-ocular, oro-tragal cleft, or oro-temporal facial cleft. This syndrome is rarely hereditary.
The main syndromes included in this group are familial facial dysplasia, Goldenhar syndrome, and congenital nonprogressive hemiatrophy of the face.
Familial Facial Dysplasia or first branchial arc syndrome presents with large deformities of the malar area and ear, and quite often is associated with anophthalmos or microphthalmos. Associated systemic defects are scoliosis of spine and hemivertebral anomalies.
Goldenhar syndrome or oculo-auricular dysplasia consists of a triad of anomalies: Epibulbar dermoids (Fig. 39.1), accessory auricular appendages, and aural fistulae. This syndrome is unilateral and associated with mandibulofacial dysostosis, hypoplasia of the jaw, coloboma of the upper lid, systemic nervous, muscular and skeletal deformities, and microphthalmos.
Congenital Nonprogressive Hemiatrophy of the Face may be another manifestation of first and second branchial developmental arrest. Patients present with hypoplasia of orbital and malar bones. Parry-Romberg syndrome is the progressive form of this anomaly.
Ectodermal Mesenchymal Induction Disorder
In ectodermal mesenchymal induction disorder (also known as cryptophthalmos syndrome), the main symptom is cryptophthalmos, which appears as a small- or normalsized subcutaneous eyeball under a large fold of skin that covers the front of the eye, orbit, and cheek. The eyeball lacks appendages, lacrimal glands, and shows no development of the optic pathway. Incomplete forms, with partially skin-covered microphthalmic eyes, have been reported. Cryptophthalmos can be associated with dyscephaly, syndactyly, and malformed genitalia. Dyscephaly includes meningocele, harelip, cleft palate, and nasal and ear deformities. Genital malformations are observed both in males and females and are associated with atresia of the anus and bladder. This anomaly is transmitted as an autosomal recessive trait.
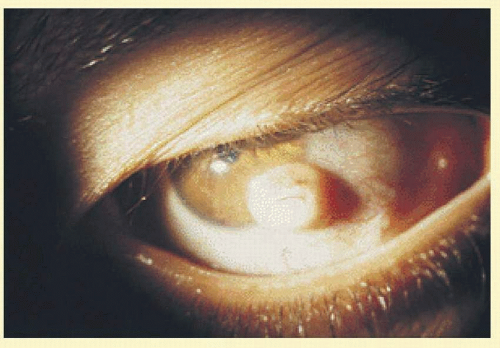 FIG. 39.1 Epibulbar dermoid with cilia. Goldenhar syndrome. (Courtesy, collection of Tel Hashomer.) |
Cranial Stenosis
Cranial stenosis is due to an aberrant development of the bones of the skull, primarily because of premature or even congenital closure of the cranial sutures. Edwards favors the classification suggested by Pemberton and Freeman as the most comprehensive classification of the different craniostenosis syndromes5 presented here.
Simple Craniostenosis may appear as scaphocephaly (boat head), due to fusion of the sagittal suture; brachycephaly (short head) due to fusion of the coronal suture; plagiocephaly (asymmetric head) due to fusion of a part of the suture; or oxycephaly (tower head), due to fusion of the coronal and another suture.
Crouzon Disease includes a brachycephalic skull, ocular proptosis due to shallow orbits, and maxillary hypoplasia. Optic nerve damage, strabismus, nystagmus, and corneal exposure also are noted. Facial features include hypertelorism, a parrot-beaked nose, nasal septum deviation, arched short palate, and irregular teeth. Deafness and atresia of the auditory meatus are also present. It is an autosomal dominant genetic disease with a wide range of expressivity.
Acrocephalosyndactyly may present as Apert syndrome, which shows irregular craniostenosis, midfacial hyperplasia, syndactyly, and shallow orbits with exophthalmus. Syndactyly is the characteristic extrafacial mark. The craniofacial appearance is similar to that of Crouzon disease. Most cases are sporadic.
Pfeiffer syndrome is another presentation of acrocephalosyndactyly in which facial features consist of a tower skull and midface hyperplasia with shallow orbits, proptosis, an antimongoloid palpebral fissure, exotropia, low-set ears, cleft palate, and dental anomalies. Syndactyly is less prominent. This disorder is autosomal dominant with complete penetrance and variable expressivity.
Carpenter syndrome, another expression of acrocephalosyndactyly, consists of a tower skull, flat nasal bridge and epicanthus, mental and growth retardation, hypogonadism, and cardiac defects. Syndactyly may be present. Obesity of the trunk and limbs helps to differentiate it from other forms of craniostenosis. It is transmitted as an autosomal recessive trait.
Miscellaneous Craniofacial Syndromes
Osteopetrosis manifests itself as osteosclerosis of the bone tissue because of a defect in bone matrix formation and bone resorption. Two syndromes are related to this defect: Pyle disease and Albers-Schönberg disease. In Pyle disease, hypertelorism and a broad, flat nasal bridge are characteristic. In Albers-Schönberg disease, macrocephaly with frontal bossing and hydrocephalus are characteristic. Both autosomal dominant and autosomal recessive heredity have been claimed in their genetic transmission.
Hallermann-Streiff Syndrome presents with a birdface profile and micrognathia, dwarfism, dental anomalies, and hypotrichosis. The main ocular symptoms are bilateral microphthalmos and cataract, blue sclera, keratoglobus, anterior segment dysgenesis, glaucoma, and coloboma. Retinal folds and optic nerve anomalies can be observed. Most cases are sporadic.
Treacher-Collins Syndrome (mandibulofacial dysostosis) presents with malar hyperplasia, receding chin, a down-slanting palpebral fissure, external ear malformation, mandibular hypoplasia, conductive deafness, macrostomia, a high palate, and teeth anomalies. The main ocular symptom is an occasional microphthalmos.
Pierre-Robin Syndrome presents with glossoptosis, micrognathia, and cleft palate. The ocular symptoms include congenital glaucoma, high myopia, esotropia, cataract, microphthalmos, uveal coloboma, and Mobius syndrome.
Rubinstein-Taybi Syndrome is associated with an antimongoloid slant of the palpebral fissure, epicanthus, ptosis, and cataract. Systemic findings include a characteristic facies with low-set ears, hypoplastic maxilla, beak nose, and characteristic broad and angulate thumbs and toes. All reported cases seem to be sporadic.
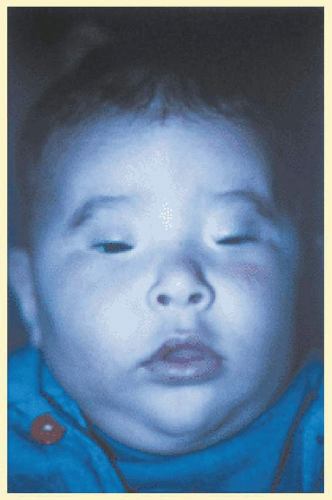 FIG. 39.2 Blepharophimosis. (Courtesy, collection of Tel Hashomer.) |
Congenital Familial Blepharophimosis is associated with epicanthus, telecanthus, ptosis, and a low nasal bridge. Structurally and functionally, the eyeballs and visual system are normal. It is an autosomal dominant disorder (Fig. 39.2).
Waardenburg Syndrome presents with lateral displacement of the median canthus, a broad and high nasal bridge, medial hyperplasia of the eyebrows, partial albinism, heterochromia iridis, and deafness. Ocular function is normal. It is an autosomal dominant disorder.
Cornelia de Lange Syndrome presents with hirsutism, a thin down-turning upper lip, micromelia, proptosis, optic atrophy, uveal coloboma, and myopia. A genetic mutation is suggested as an etiologic factor.
Cockayne Syndrome is associated with senile aging starting after 1 to 2 years of normal growth and a progressive retinal degeneration.5
Ocular Anomalies Observed in Drug Embryopathies
Ocular anomalies appear in children affected with drug embryopathies.8
Fetal Alcohol Syndrome
Fetal alcohol syndrome is caused by excessive ingestion of alcohol by pregnant women. This syndrome presents with mental retardation and numerous cristopathies. The main systemic findings are midface hypoplasia, a flat nasal bridge and thin upper lip, microcephalus, hyperactivity, and seizures.8 Among the ocular findings, the external signs are short palpebral fissures, epicanthus, ocular hypertelorism and telecanthus, coloboma of the iris, strabismus, blepharoptosis, and microphthalmos. The internal signs are hypoplasia of the optic nerve, increased tortuosity of the retinal vessels, and a disturbed retinal function on the basis of abnormal electroretinogram.9 The refractive status shows a great variability from -23 diopters of myopia to +6,5 diopters of hypermetropia.10
Opiates Embryopathy
Heroin and methadone are the chief opiates involved in fetal exposure. As opiate withdrawal can be more harmful to the fetus than continued drug exposure, no efforts are made to wean methadone-dependent women during pregnancy. Withdrawal (Neonatal abstinence syndrome NAS) of methadone causes a developmental delay, delayed visual maturation with reduction in visual acuity, and nystagmus.11
Cocaine Embryopathy
Cocaine can easily access the fetal circulation and causes a developmental delay and structural brain abnormalities such as Septo-optic dysplasia, porencephaly, and bilateral cortical infarcts. There is a vascular disruption in the retina manifesting itself with superficial and deep retinal hemorrages, a delayed visual maturation, and optic nerve abnormalities.11
Thalidomide Embryopathy
Thalidomide taken during the first trimester of pregnancy causes phocomelia (shortened limbs). Ocular signs are Duane and Mobius syndromes, microphthalmos, uveal coloboma, and, occasionally, anophthalmos.8
Lysergic Acid Diethylamide Embryopathy
Lysergic acid diethylamide (LSD) taken during the first trimester of pregnancy can cause arhinencephaly, a fusion of the frontal lobes, hydrocephalus with Arnold-Chiari syndrome, and an absence of convolutions on the brain. Ocular signs include microphthalmos, cataract, and retinoschisis.8
Fetal Anticonvulsant Syndrome
Prenatal exposure to anticonvulsant drugs either as Valproate monotherapy or in combination with other drugs is the cause of the fetal anticonvulsant syndrome. It is characterized by the presence of epicanthic folds, medially deficient eyebrows, shallow nasal bridge with anteverted nares and trigonocephaly. There is a high incidence of myopia and strabismus.11
Antiangiogenic Agents As a Cause of Embryopathy
Maternal administration of angiogenic inhibitors TN-470 and Angiostatin 4,5 cause fetal growth restriction and placental abnormalities, which in turn induce microphthalmos.12
Coumarin Embryopathy
In coumarin embryopathy the infant presents with impaired coagulation, intraventricular and parenchymal minor cerebral hemorrhages, mid-face hypoplasia, hepatopathy with elevated liver enzymes, and episodes of hypoglycemia. The ocular symptoms are corneal opacities with anterior segment dysgenesis, persistent pupillary membrane, cataract, and persistent primary hyperplastic vitreous.13
Ocular Anomalies Due to Infectious Embryopathies
Congenital Rubella Syndrome
Congenital rubella syndrome consists of cataract, congenital glaucoma, iris abnormalities, and secondary pigmentary retinopathy. Systemic findings include cardiovascular defects, mental retardation, deafness, central nervous system (CNS) abnormalities, thrombocytopenic purpura, diabetes mellitus, osteomyelitis, dental abnormalities, pneumonitis, hepatomegaly, and genitourinary anomalies. The rubella virus passes through the placenta, infects the fetus, and causes abnormal embryogenesis. The rubella virus can survive in the lens for at least 3 years after birth. Surgical section of the rubella cataract may release the virus and cause an endophthalmitis.14
Cytomegalovirus Embryopathy
Cytomegalovirus (CMV) infection is the most common known viral infection acquired in utero. It is an important cause of fetal demise and intrauterine growth retardation. Ninety percent of congenitally infected infants are asymptomatic. But in infected infants, congenital CMV is a major cause of developmental and neurologic abnormalities.15 Children with congenital CMV infection following first trimester maternal infection are more likely to have CNS sequelae than those whose mothers were infected late in pregnancy. The main CNS signs are secondary hearing loss, mental retardation, cerebral palsy, and seizures. The main ocular symptom is chorioretinitis.16
Congenital Varicella Syndrome
The varicella-zoster virus (VZV), when active during the first and second trimester of pregnancy, causes an embryopathy or fetopathy called Congenital Varicella Syndrome (CVS) in 25% of the cases. The characteristic symptoms consist of skin lesions in dermatomal distribution, neurologic defects, eye diseases, and limb hypoplasia. The skin lesions appear as areas of scar or skin loss. The neurologic defects are cortex and spinal atrophy, encephalitis, microcephaly, limb paresis, seizures, dysphagia, and Horner’s syndrome. The ocular symptoms are microphthalmos, enophthalmos, anisocoria, chorioretinitis, optic atrophy, cataract, and nystagmus. In addition, skeletal abnormalities, muscle hypoplasia, gastrointestinal and genitourinary abnormalities, cardiovascular defects, and developmental delay are observed.17
Herpes Simplex Virus Embryopathy
Herpes simplex virus (HSV) disease of the newborn is acquired mostly during the perinatal period. The in utero affected infant presents a triad of manifestations: cutaneous (scarring, active lesions, hypo- and hyperpigmentation, aplasia cutis, erythema), ophthalmic (microphthalmos, retinal dysplasia, optic atrophy, chorioretinitis), and neurologic (microcephaly, encephalomalacia, hydranencephaly, intracranial calcification). Clinical manifestations in the CNS include seizures, lethargy, irritability, tremor, and temperature instability.18
Human Immunodeficiency Virus and the Neonate
Transmission of HIV from mother to infant is not absolute and happens in 13% to 39% of infants. HIV can affect all the organ systems, and its manifestations can be infectious and noninfectious. Pediatric AID patients suffer often from infections caused by common bacterial and viral (HSV, VZV, CMV) pathogens. Noninfectious complications include dermopathies, progressive neurologic disease, LIP disease (diffuse infiltration of the pulmonary alveoli by lymphocytes and plasma cells), hematologic dyscrasias, renal, gastrointestinal, and cardial and ocular complications. Dermatologic manifestations include common childhood skin infections, atrophy of the skin and nails, and drug hypersensitivity. Hematologic manifestations are anemia, leucopenia, and thrombocytopenia. Renal manifestations are due to glomerulopathy. The gastrointestinal symptoms are recurrent aphthous ulcers in the mouth, esophagitis, hepatitis with cholelithiasis, and pancreatitis. The main cardial symptom is cardiomyopathy. Ocular manifestations are not specific for the HIV: simple eye infections such as blepharitis and conjunctivitis are common. The severe retinopathies seen in adults are less common in infants. However, children with retinitis present a bilateral involvement and complain of a decrease in the central and peripheral vision.19
Toxoplasmosis Embryopathy
The embryopathy caused by these protozoa involves mostly the CNS and the eye. The CNS lesions include hydrocephalus, microcephalus, focal cerebral calcification, and occasionally meningoencephalitis associated with mental retardation, spasticity, and palsies. The eye is mostly the site of retinochoroiditis with a predilection for the macula, and when it harbors an active inflammatory process, it is the site of iridocyclitis, vitreitis associated with traction retinal detachment, and papillitis. Also one can observe cataract, microphthalmos, nystagmus, and strabismus. The involvement of the other organs presents with hepatomegaly, splenomegaly, thrombocytopenia, anemia, nephritic syndrome, and deafness.20
Congenital Syphilis
Congenital syphilis is the cause of a bilateral, stromal keratitis that is associated with the triad of Hutchinson: Hutchinson teeth, keratitis, and deafness.21
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


