appear to play a role and genetic abnormalities are not yet delineated. In Sturge-Weber syndrome, there is some evidence that the condition is a mosaic mutation with only segmental involvement. Those that receive the full mutation die in utero. Although these conditions are generally hereditary, many do not become clinically apparent until the teenage years or young adulthood.
bodies, are frequently present in the tumor. The uveal tract is rarely affected in tuberous sclerosis. A depigmented iris sector, seen in some affected patients, is believed to be the equivalent of depigmented cutaneous lesions.
Table 20.1 DIAGNOSTIC CRITERIA (REVISED) FOR ESTABLISHING THE DIAGNOSIS OF TUBEROUS SCLEROSIS COMPLEX (TSC) | ||||||||
|---|---|---|---|---|---|---|---|---|
|
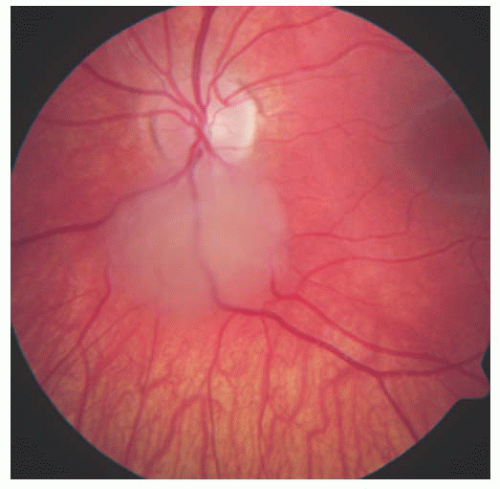 FIGURE 20.1. Tuberous sclerosis complex: non-calcified retinal astrocytic hamartoma. |
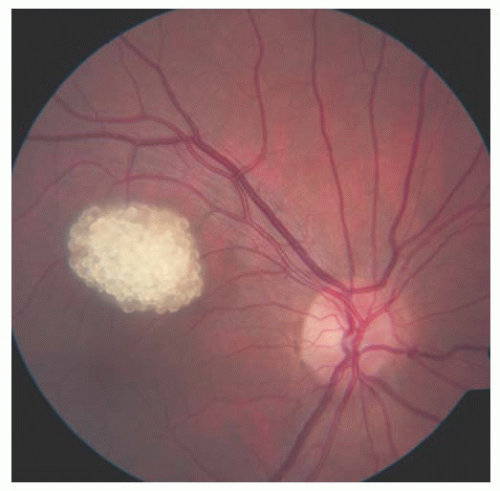 FIGURE 20.2. Tuberous sclerosis complex: calcified retinal astrocytic hamartoma. |
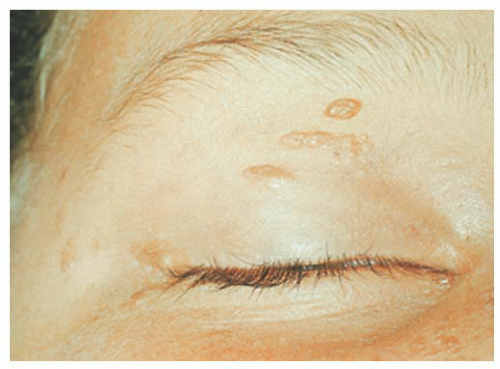 FIGURE 20.3. Tuberous sclerosis complex: adenoma sebaceum in the periocular region. |
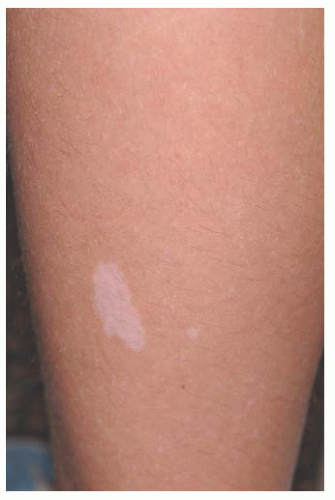 FIGURE 20.4. Tuberous sclerosis complex: ash leaf sign. |
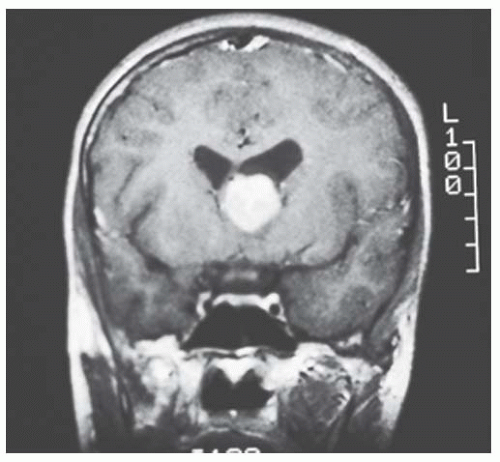 FIGURE 20.5. Tuberous sclerosis complex: brain astrocytoma. |
or to metastasize (26). The cardiac lesions have been shown to be rhabdomyomas, composed of large spider cells with prominent vacuoles containing glycogen. Some patients with tuberous sclerosis develop slowly progressive subpleural cysts that result from anomalous development of pulmonary tissue. These cysts can rupture, leading to spontaneous pneumothorax. Irregular cortical thickenings of bones, particularly the metatarsals and metacarpals, as well as hamartomas of the liver, thyroid, pancreas, testes, and other organs, have been reported.
elements. There appears to be a higher incidence of uveal melanoma in patients with neurofibromatosis.
Table 20.2 DIAGNOSTIC CRITERIA FOR NEUROFIBROMATOSIS TYPE 1 (NF1) | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
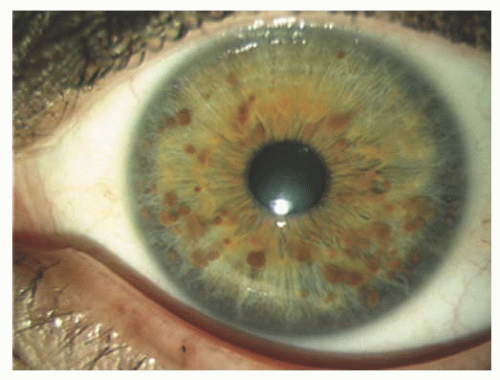 FIGURE 20.6. Neurofibromatosis type 1: Iris Lisch nodules. |
or subtle features of color or visual acuity abnormalities (Figs. 20.8 and 20.9). Magnetic resonance imaging shows enlargement of the optic nerve, often so large that it develops a fold (kink) within its substance to accommodate the orbit, leading to down-and-out proptosis. This mass shows enhancement on T1-weighted, gadolinium contrast images, particularly notable in the axial and coronal views. It is important to differentiate this tumor from optic nerve sheath meningioma as the systemic implications and therapy differ. This is best determined using gadolinium-enhanced, orbital fat suppressed, T1-weighted coronal views. With glioma, the central substance of the nerve enhances, whereas with meningioma the peripheral encircling arachnoid sheath enhances. Glioma is more often associated with neurofibromatosis type 1, whereas meningioma with neurofibromatosis type 2.
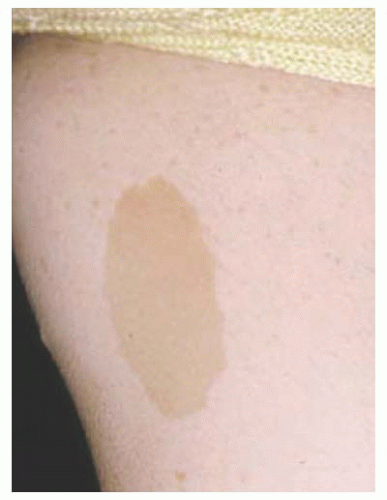 FIGURE 20.7. Neurofibromatosis type 1: cutaneous café au lait spot. |
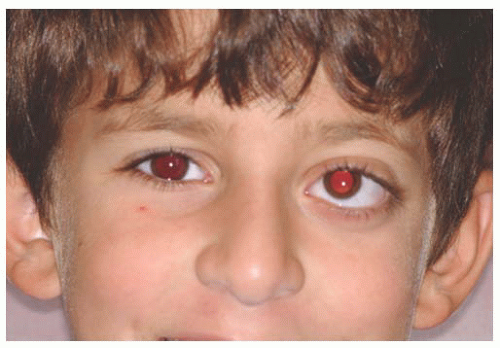 FIGURE 20.8. Neurofibromatosis type 1: mild proptosis from optic nerve glioma. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


