Pattern
Entity
Conjunctival features
Associated features
Locus/gene
Pigmentation
Carney complex
Conjunctival pigmentation
Spotty mucocutaneous pigmentation
17q
Schwannoma
Endocrine over-activity
PRKAR1A gene chromosome 2
Testicular tumor
Peutz–Jeghers syndrome
Conjunctival pigmentation
Mucocutaneous pigmentation Gastrointestinal polyposis
19p13.3
STK11
Benign tumors
Dermoid
Organoid nevus syndrome
Epibulbar dermoid
Cutaneous sebaceus nevus
Sporadic
Coloboma
Goldenhar syndrome
Epibulbar dermoid
Preauricular appendages
Sporadic
Pretragal fistula
Vertebral anomalies
Proteus
Epibublar dermoid
Connective tissue nevi
Sporadic
Strabismus
Lipoma
Orbital exostoses
Vascular malformations
Epidermal nevi
Neuroma
MEN 2B
Conjunctival neuroma
Thickened corneal nerves
10q11.2
Mucocutaneous neuroma
RET proto-oncogene
Endocrine tumor
Malignant tumors
Xeroderma pigmentosum
Conjunctiva xerosis
Skin atrophy with pigmentary changes
Variable
Keratitis
Neurological abnormalities
Ocular surface neoplasms
Amyloidosis
Conjunctival nodule
Variable
Sporadic
Conjunctival hemorrhage
Familial
22.1 Carney Complex
Carney complex is characterized by cutaneous pigmentary abnormalities, myxomas, endocrine tumors, and schwannomas [1]. Conjunctival and caruncular pigmentation may be present in about one-fourth of cases [2]. Similar pigmentation of the skin is also typically distributed in the lips and genital mucosa [3] (see Chaps. 2, 3, 4, 5, 6, 7, 8, 9, 10, and 11 for further details).
22.2 Peutz–Jeghers Syndrome
Peutz–Jeghers syndrome refers to the association of gastrointestinal hamartomatous polyposis and mucocutaneous pigmentation [4, 5].
22.2.1 Inheritance
Peutz–Jeghers syndrome is inherited in an autosomal dominant pattern.
22.2.2 Molecular Genetics
Mutations of STK11, a tumor suppressor gene on chromosome 19p13.3, are detectable in about 20–70 % of patients [6, 7].
22.2.2.1 Ophthalmic Features
Congenital hypertrophy of the retinal pigment epithelium (CHRPE)-like lesions of the fundus that are characteristic of patients with Gardner syndrome does not occur in Peutz–Jeghers syndrome [8]. However, pigmented spots of the eyelids and conjunctiva have been observed (Fig. 22.1) [9].
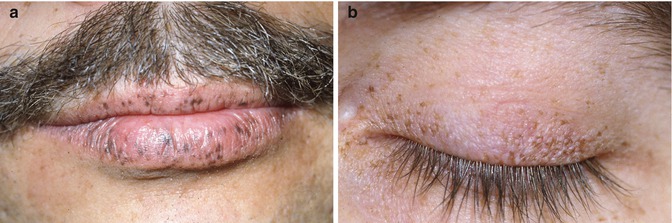
Fig. 22.1
Pigmentation of lips. (a) and eyelid (b) seen in Peutz-Jeghers syndrome
22.2.3 Systemic Features
Perioral pigmentation is pathognomonic, particularly if it occurs across the vermilion border. Oral mucosa and fingertips are also commonly affected. About 50 % of patients develop a wide variety of cancers in adulthood [10]. An overexpression of COX-2 has been reported in polyps and cancers related to PJS [11].
22.3 Sebaceous Nevus Syndrome
Sebaceus nevus syndrome (of Jadassohn), also known as Schimmelpenning–Feuerstein–Mims syndrome, is a distinct clinical disorder within the spectrum of epidermal nevus syndrome (of Solomon) characterized by cutaneous sebaceus nevus and extracutaneous manifestations [12]. The classic triad involves facial lesions, seizures, and mental retardation [13]. Ocular involvement is observed in about 40 % of cases with epibulbar choristomas and coloboma of the eyelid being the most common. The limbal choristomas can be simple or complex and usually are dermoid or lipodermoid in nature. (See Chaps. 5, 6, 7, and 8 for further discussion.)
22.4 Goldenhar Syndrome (Oculoauriculovertebral Dysplasia)
Goldenhar described a triad of epibulbar dermoids, preauricular appendages, and pretragal fistula [14]. Since then, the spectrum of manifestations has expanded to include vertebral anomalies and is also called oculoauriculovertebral dysplasia (OAV) [15]. It is now considered as a specific subtype of hemifacial microsomia which is associated with epibulbar dermoids. The etiology of GS has yet to be elucidated, but it has been suggested that a vascular insult and/or neural crest abnormality during embryogenesis could account for this syndrome [16].
22.4.1 Inheritance
The majority of cases occur sporadically. Exceptional cases with autosomal dominant and recessive inheritance patterns have also been reported [17].
22.4.2 Molecular Genetics
22.4.3 Ophthalmic Features
Epibulbar dermoid is a necessary feature for the diagnosis of Goldenhar syndrome (Fig. 22.2). The epibulbar dermoids are almost always located near the limbus in the inferotemporal quadrants. Secondary irregular astigamstism and anisometropic amblyopia may also be present. Dermolipomas are typically in the subconjunctival plane and appear as a yellow soft mass in the superotemporal quadrant. Other associated anomalies include eyelid coloboma [20], Duane syndrome [21], and caruncular anomalies [22]. Involvement of the globe itself manifesting as microphthalmia and coloboma is uncommon [23].
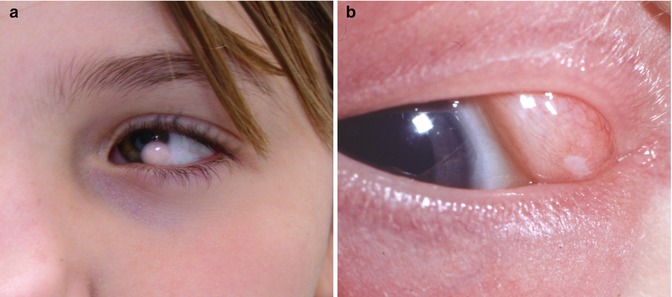
Fig. 22.2
Epibulbar dermoids in Goldenhar syndrome. Limbal (a) and extralimbal (b)
22.4.4 Systemic Features
In addition to ophthalmic, ear, and vertebral anomalies, about 50 % of cases have anomalies such as micrognathia, macrostomia, cleft lip and palate, and developmental defects of the heart [24]. Recent analysis of data suggests a statistical association with the VATER anomaly (vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, and radial dysplasia) and CHARGE syndrome (coloboma, heart anomaly, choanal atresia, retardation, genital and ear anomalies) [25].
22.5 Proteus Syndrome
Proteus syndrome is a severe and highly variable disorder characterized by asymmetric and disproportionate overgrowth of body parts and hamartomas, along with a susceptibility to tumors [26]. Proteus syndrome was first recognized as a specific entity in 1979 [27] and named after the Greek god Proteus, as he could change his shape, emphasizing the varied manifestations of the syndrome [28].
22.5.1 Inheritance
22.5.2 Molecular Genetics
22.5.3 Ophthalmic Features
Ophthalmic involvement is common in Proteus syndrome. A review of more than 200 published cases revealed that more than 40 % of cases that met the diagnostic criteria had one or more ophthalmic features [32]. Epibulbar and eyelid dermoids, strabismus, nystagmus, high myopia, orbital exostoses, and posterior segment hamartoma are most commonly observed [33–35]. A complete list of published ophthalmic findings is given elsewhere [32–34].
22.5.4 Systemic Features
The manifestations are usually present at birth and progress during childhood. Due to its varied manifestations, the diagnosis of Proteus syndrome is frequently missed. Critical review of published cases revealed that only 47 % of cases met the diagnostic criteria [32]. General features suggestive of this diagnosis include mosaic distribution of lesions, a progressive course, and sporadic occurrence [32]. Connective tissue nevi are pathognomonic. Other features include lipoma (92 %), vascular malformations (88 %), and epidermal nevi (67 %) (Fig. 22.3) [30]. The list of diagnostic criteria is reviewed elsewhere [36]. Individuals with significant clinical features but who do not meet the diagnostic criteria are labeled as having Proteus-like syndrome. The differential diagnosis of Proteus syndrome includes neurofibromatosis type 1, Klippel–Trenaunay–Weber syndrome, Maffucci’s syndrome, Ollier’s disease, and Bannayan–Ruvalcaba–Riley syndrome.
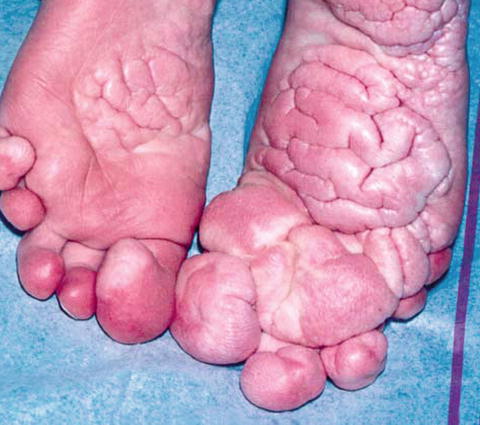
Fig. 22.3
Cerebriform connective tissue nevi of both soles are characteristic of Proteus syndrome (Reproduced with permission from Nguyen et al. [30])
22.6 Multiple Endocrine Neoplasia Type 2B
Multiple endocrine neoplasia (MEN) refers to a genetic predisposition to develop benign and malignant tumors of various endocrine glands. Based upon the pattern of glandular involvement, MEN is classified as type 1 and type 2. MEN type 2 is comprised of three subtypes: familial medullary thyroid carcinoma, MEN 2A, and MEN 2B. MEN 2B, also called mucosal neuroma syndrome or Wagenmann–Froboese syndrome, is the only MEN subgroup that is of ophthalmic interest because of its association with mucosal neuromas and corneal abnormalities [37, 38].
22.6.1 Inheritance
MEN 2B is inherited as an autosomal dominant trait. About half the cases are due to de novo mutations.
22.6.2 Molecular Genetics
More than 95 % of patients with MEN 2B have a point mutation in the RET gene [39].
22.6.3 Ophthalmic Features
A comprehensive review of all published cases of MEN 2B reveals that common ophthalmic manifestations include prominent corneal nerves (100 %), eyelid neuroma or thickening (88 %), subconjunctival neuroma (79 %), and dry eyes (48 %) (Fig. 22.4) [40–42]. With rare exceptions [43], the presence of prominent corneal nerves in otherwise normal cornea should lead to investigations to exclude MEN 2B [40, 42]. Other infrequent causes of prominent corneal nerves such as neurofibromatosis, leprosy, and congenital ichthyosis should also be considered in the differential diagnosis [42]. Histopathologic findings of the cornea have shown that prominent corneal nerves are axonal bundles of nonmyelinated nerves in association with Schwann cells [44, 45].
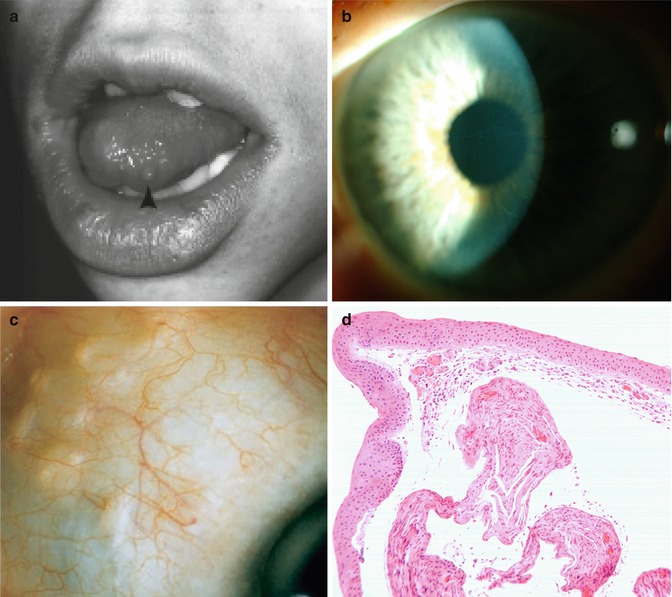
Fig. 22.4
Characteristic features of MEN 2B syndrome. Submucosal lip and tongue neuroma (a, arrow) (Reproduced with permission from Jacobs and Hawes [40]). Prominent corneal nerves (b). Plexiform subconjunctival neuroma (c) (Reproduced with permission from Eter et al. [41]). Histopathologic section of the conjunctiva shows thickened abnormal nerves in the substantia propria (d)
22.6.4 Systemic Features
A clinical diagnosis of MEN 2B is suspected in the presence of a marfanoid body habitus, mucosal neuromas of the lips and tongue, prominent corneal nerves, and medullary thyroid carcinoma [38]. As medullary thyroid carcinoma tends to be aggressive, early prophylactic thyroidectomy is recommended [46]. Patients may also have chronic constipation and colonic cramping due to megacolon disorder [47]. About half of patients with MEN 2B also develop pheochromocytoma. Unlike other types of MEN, involvement of the parathyroid gland is rare in MEN 2B [48].
22.7 Xeroderma Pigmentosum
Xeroderma pigmentosum is a genetic disorder characterized by extreme sensitivity to sunlight and a constellation of cutaneous, ophthalmic, and neurological findings [49]. It is rarely seen in Europe and North America and is relatively more common in Japan, North Africa, and the Middle East.
22.7.1 Inheritance
22.7.2 Molecular Genetics
Xeroderma pigmentosum is due to mutations of one of eight genes that are involved in nucleotide excision repair [51–53]. The products of seven of these genes (XP–A through G) are involved in nucleotide excision repair of DNA damaged by UV light [50]. The eighth gene (POLH) codes for a special polymerase used to replicate damaged DNA [50]. Cockayne syndrome and trichothiodystrophy, two related disorders of defective nucleotide excision repair mechanisms [54], unlike xeroderma pigmentosum, are not associated with the risk of developing skin cancers [55].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


