Entity
Eyelid tumor
Associated features
Locus/gene
Neurofibromatosis type 1
Neurofibroma
Lisch nodules
17q
Café au lait spots
NF 1 gene
Pheochromocytoma
Sturge-Weber Syndrome
Diffuse hemangioma
Leptomeningeal hemangioma
Sporadic
Gardner syndrome
Epidermoid cyst
CHRPE
5q21
Fibroma
Colorectal polyps/carcinoma
APC gene
Orbital osteoma
Gorlin-Goltz syndrome
Basal cell carcinoma
Odontogenic cysts
9q 22
Bifid ribs
PTC gene
Palmar pits
Ovarian tumor
Cowden syndrome
Trichilemmoma
Oral papilloma
10q23
Breast tumor
PTEN gene
Thyroid tumor
Carney complex
Myxoma
Spotty mucocutaneous pigmentation
17q
Schwannoma
PRKAR1A gene chromosome 2
Endocrine overactivity
Testicular tumor
Muir-Torre syndrome
Sebaceous adenoma
Keratoacanthoma
2p
Basal cell carcinoma
hMLH1, hMSH2
Colorectal adenocarcinoma
Patients with an inherited predisposition for tumors tend to develop these tumors at an earlier age, have multiple tumors with bilateral involvement, and may have a positive family history of similar lesions [1]. The majority of eyelid tumors in the setting of an inherited predisposition are benign, but some malignant tumors are also known to have a syndromic association such as ones that occur, for example, in the setting of the Gorlin-Goltz syndrome. One of the most important clues to the presence of an associated systemic disease is an unusual histopathologic feature of the tumor for its location in the eyelid. For example, tumors such as myxomas or sebaceous adenomas of the eyelid are unlikely to occur in absence of a syndromic association. Eyelid tumors can also be metastatic, but such cases are rare and account for less than 1 % of malignant eyelid tumors [2].
12.1 Neurofibroma
Neurofibroma is the hallmark finding of neurofibromatosis type 1 (NF1). NF1 is inherited in an autosomal-dominant fashion with a nearly even split between spontaneous and inherited mutations [3]. The disease is due to mutations in the NF1 gene, which is located on chromosome 17q11.2 and encodes for the protein neurofibromin [3]. Neurofibromas tend to be multiple and develop towards the end of the first decade of life, with penetrance approaching 100 % by the second decade of life [3]. Expressivity is highly variable, even among family members [3]. The neurofibromas appear as discrete soft tumors on the face (including eyelids), hands, and trunk. Based on their appearance and extent of tissue involvement, neurofibromas can be classified as cutaneous, subcutaneous, nodular plexiform, and diffuse plexiform. Often, neurofibromas of the eyelids can lead to ptosis and sometimes detachment of the lateral canthal ligament. Neurofibromas in the upper eyelid can also lead to an “S”-shape deformity because the tumor infiltrates more the lateral side of the lid [4].
12.2 Nevus Flammeus
In general, only about 10 % of all patients with nevus flammeus or port-wine stain of the eyelid are associated with Sturge-Weber syndrome (SWS) (Fig. 12.1) [5]. Sturge-Weber syndrome only occurs in patients who have hemangiomas in the V1 or V2 areas of distribution of the trigeminal nerve. Bilateral port-wine stains of the eyelids have a higher likelihood of being associated with Sturge-Weber syndrome than unilateral lesions [5]. In a study of 55 patients with Sturge-Weber syndrome, glaucoma was the main ocular disease and was related to the extension of the nevus flammeus in the palpebral area [6]. Patients in the study who did not develop a nevus flammeus in the palpebral area did not develop glaucoma or choroidal hemangioma [6]. In the absence of leptomeningeal involvement, patients should only be given a diagnosis of nevus flammeus, port-wine stain, or facial angioma to avoid the stigmata associated with a diagnosis of Sturge-Weber syndrome.
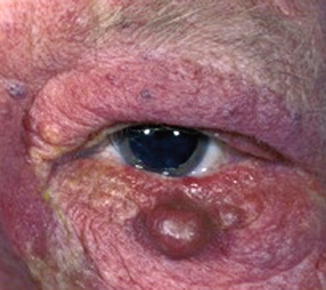
Fig. 12.1
Sturge-Weber syndrome. In addition to typical diffuse cutaneous involvement, note nodular hemangioma
12.3 Gardner Syndrome
Gardner syndrome was first described by Eldon J. Gardner in 1951 as a autosomal-dominant disorder characterized by the triad of multiple osteomas, colonic polyposis, and benign tumors of the skin and soft tissues [7]. Gardner syndrome is a variant of familial adenomatous polyposis caused by a mutation in the APC gene on chromosome 5q21 [7]. Extracolonic manifestations of Gardner syndrome include radiologic jaw anomalies, pigmented ocular fundus lesions that resemble congenital hypertrophy of the retina pigment epithelium, soft tissue tumors, desmoid tumors, and other cancers [8, 9]. Orbital osteoma, soft tissue tumors of the brows or eyelids, and epidermoid cysts of the eyelid may also occur [10–12].
12.4 Nevoid Basal Cell Carcinoma Syndrome (Gorlin-Goltz Syndrome)
Nevoid basal cell carcinoma syndrome (NBCCS) is also referred to as basal cell nevus syndrome, Gorlin syndrome, or Gorlin-Goltz syndrome [13, 14]. Although basal cell nevi may occur in early childhood, it is the risk for multiple basal cell carcinoma and developmental anomalies that characterizes NBCCS [15]. Other features include development of multiple jaw keratocysts and characteristic facial features.
12.4.1 Inheritance
NBCCS is inherited as an autosomal-dominant trait with complete penetrance and variable expressivity [16]. About 30 % of probands have a de novo mutation.
12.4.2 Molecular Genetics
This disorder is due to mutations in the PTCH gene on chromosome 9q22.3 [17, 18]. PTCH sequence alterations can be detected in about 60–85 % of cases that meet clinical diagnostic criteria for NBCCS [18]. Studies have shown that the PTCH gene may be inactivated in the standard two-hit hypothesis, by haploinsufficiency or through dominant-negative isoforms [19].
12.4.3 Ophthalmic Features
A wide variety of ophthalmic manifestations may be present in 26 % of patients with NBCCS [15]. Periocular basal cell carcinoma [20, 21], hypertelorism, nystagmus, and cataracts are some of the common features (Fig. 12.2) [15, 21]. The occurrence of microphthalmia [22], coloboma, combined retinal and RPE hamartoma [23], and vitreoretinal abnormalities [24] in the setting of NBCCS implicates the PTCH gene in ocular development [22, 24].
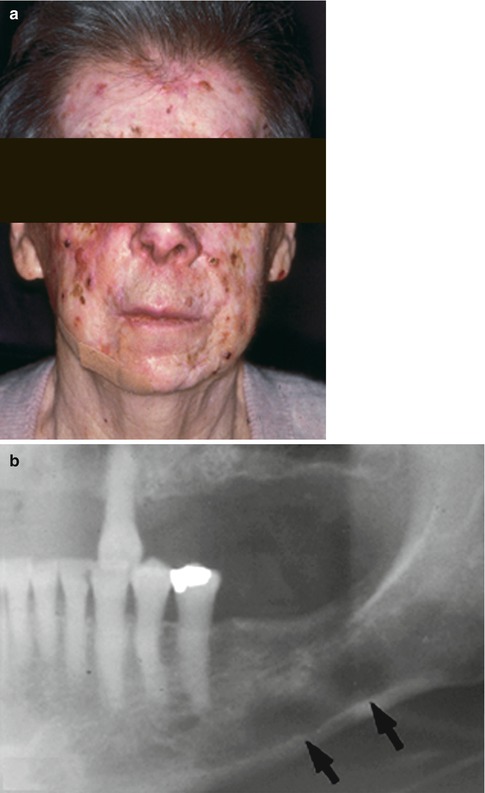
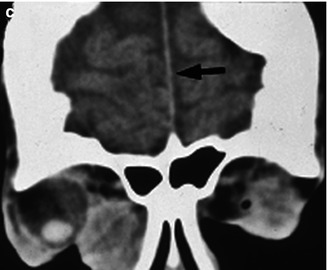
Fig. 12.2
External photograph showing multiple facial basal cell carcinoma (a). Orthopantomograph of the right mandible shows odontogenic keratocysts seen as round, well-circumscribed radiolucent areas (b, arrows). Coronal noncontrast computed tomography scan of the skull showing of falx cerebri (arrow) and large diffuse lesion in the medial orbit (c) (Reproduced with permission from Honavar et al. [21])
12.4.4 Systemic Features
Jaw keratocysts and calcification of the falx cerebri are some of the most frequent (90 %) manifestations of the NBCCS (Fig. 12.2) [15, 25]. Basal cell carcinomas tend to be multiple (>5 in a lifetime) and occur before the age of 30 years. They may arise from preexisting basal cell nevi or de novo. In general, about 0.5 % of patients with basal cell carcinoma have underlying NBCCS [26]. The proportion is much higher (22 %) in patients with basal cell carcinoma prior to age 20 years [27]. White race, sun exposure, and radiation therapy are major risk factors for inducing basal cell carcinoma [28]. The characteristic facial features include forehead bossing and macrocephaly [25]. The diagnostic criteria for NBCCS are summarized in Table 12.2 [15]. Patients with NBCCS are expected to have a normal life expectancy [15].
Table 12.2
Clinical diagnostic criteria for nevoid basal cell nevus syndrome
Major criteria | ≥2 major and ≥1 minor criteria | Jaw keratocyst |
Falx calcification | ||
Palmar or plantar pits | ||
Basal cell carcinoma >5 in a lifetime or before the age of 30 years | ||
Affected first-degree relative | ||
Minor criteria | ≥1 major and ≥ 3 minor criteria | Macrocephaly |
Medulloblastoma | ||
Lympho-mesenteric or pleural cysts | ||
Cleft lip/palate | ||
Vertebral anomalies | ||
Polydactyly | ||
Ovarian/cardiac fibromas | ||
Ocular anomalies |
12.5 Multiple Hamartoma Syndrome (Cowden Syndrome)
Cowden syndrome was first described in 1963 and is named after the surname of the patient on whom the initial observations were made [29]. As the majority of tumors in this syndrome are hamartomatous malformations, it has also been referred to as multiple hamartoma syndrome [30]. Other features include benign and malignant tumors of the thyroid, breast, and endometrium [30].
12.5.1 Inheritance
12.5.2 Molecular Genetics
Approximately 80 % of individuals who meet the clinical diagnostic criteria have detectable PTEN (phosphatase and tensin homolog) missense mutations [33–35]. PTEN is a tumor suppressor gene found on chromosome 10 that acts as a lipid phosphatase in the PI3K/Akt pathway to arrest the cell in G1 phase and promote apoptosis [35]. PTEN also acts as a protein phosphatase in the MAPK pathway which regulates cell survival [35]. Finally, PTEN has been hypothesized to play a role in cell migration and adhesion due to its homology to focal adhesion molecules such as tensin and auxilin [35]. Identification of a PTEN mutation is necessary to make the diagnosis of Cowden syndrome. As PTEN mutations are also present in closely related clinical entities, Cowden syndrome is now considered within the spectrum of the PTEN hamartoma tumor syndrome which includes Bannayan-Riley-Ruvalcaba syndrome, Proteus syndrome, and Proteus-like syndrome [34].
12.5.3 Ophthalmic Features
Eyelid trichilemmomas are hallmark manifestation of Cowden syndrome (Fig. 12.3).
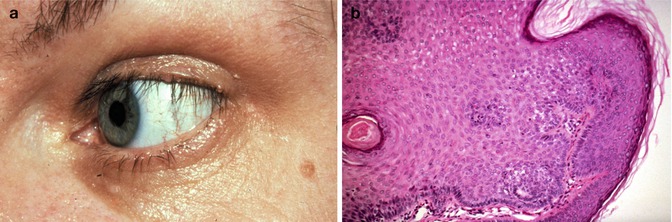
Fig. 12.3
Clinical photograph of flesh-colored popular lesions at the eyelid margin (a). High-power photomicrograph of trichilemmoma with basal palisading and bland-looking cells with more cytoplasm than basal cell carcinoma cells (b, H&E original magnification ×200) (Reproduced with permission from et al. [36])
12.5.4 Systemic Features
In a review of published cases, the age of onset ranged from 4 to 75 years [38]. Mucocutaneous lesions such as trichilemmomas, papillomatous papules, acral keratoses, and plantar keratoses are most striking manifestations. More significantly, Cowden syndrome is associated with a lifetime increased risk for breast tumors (benign 67 %, malignant 25–50 %) [39], thyroid tumors (benign 75 %, malignant 10 %) [40], and uterine tumors (benign fibroids and malignant 10 %). Other uncommon hamartomatous manifestations include gastrointestinal polyps and cerebellar dysplastic gangliocytoma (Lhermitte-Duclos disease) [34].
Consensus diagnostic criteria include pathognomonic, major, and minor criteria based upon which a clinical diagnosis of Cowden syndrome is made (Table 12.3) [31]. However, identification of a PTEN mutation is necessary to establish the diagnosis. The presence of multiple (three or more) trichilemmomas among other mucocutaneous manifestations should raise a strong suspicion of Cowden syndrome [31].
Table 12.3
International Cowden Consortium criteria for clinical diagnosis
Pathognomonic | Adult cerebellar dysplastic gangliocytoma | Operational diagnosis |
Trichilemmomas (facial) | ≥6 facial papules, of which ≥3 are trichilemmoma | |
Acral keratoses | Facial papules and oral papillomatosis | |
Papillomatous lesions | Acral keratoses and oral papillomatosis | |
Mucosal lesions | ≥6 palmoplantar keratoses | |
Major | Breast cancer | ≥2 major criteria |
Thyroid cancer (non-medullary) | ||
Macrocephaly | One major and ≥3 minor criteria | |
Endometrial carcinoma | ||
Minor | Other thyroid lesions | ≥4 minor criteria |
Mental retardation | ||
Hamartomatous intestinal polyps | ||
Fibrocystic disease of the breast | ||
Lipomas | ||
Fibromas < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|