Autosomal recessive condition caused by a deficiency of the enzyme ceruloplasmin
 Onset under age 40 years
Onset under age 40 years
Symptoms
 Usually no ocular symptoms
Usually no ocular symptoms
 May experience cirrhosis, renal disease, or neurologic dysfunction (typically motor disorders)
May experience cirrhosis, renal disease, or neurologic dysfunction (typically motor disorders)
Signs
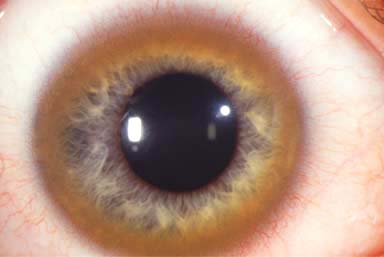
 Kayser-Fleischer ring: green-brown band of copper deposition in the peripheral zone of Descemet’s membrane (Fig. 8-1; see also Fig. 6-8). This ring usually begins in the vertical meridians and may be seen earliest with gonioscopy. A Kayser-Fleischer ring may be the presenting sign of the disease.
Kayser-Fleischer ring: green-brown band of copper deposition in the peripheral zone of Descemet’s membrane (Fig. 8-1; see also Fig. 6-8). This ring usually begins in the vertical meridians and may be seen earliest with gonioscopy. A Kayser-Fleischer ring may be the presenting sign of the disease.
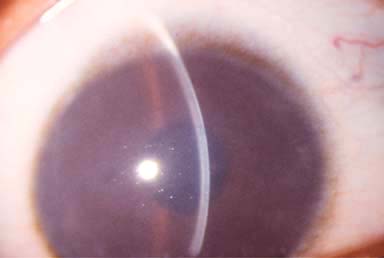
 Cataract is found in less than 10% of cases. A disc-shaped, central, polychromatic opacity with peripheral radiations can be seen (sunflower cataract).
Cataract is found in less than 10% of cases. A disc-shaped, central, polychromatic opacity with peripheral radiations can be seen (sunflower cataract).
Differential Diagnosis
 Other causes of a Kayser-Fleischer ring: primary biliary cirrhosis, chronic active hepatitis, multiple myeloma
Other causes of a Kayser-Fleischer ring: primary biliary cirrhosis, chronic active hepatitis, multiple myeloma
 Chalcosis: corneal copper deposition from an intraocular copper foreign body
Chalcosis: corneal copper deposition from an intraocular copper foreign body
Diagnostic Evaluation
 Slit-lamp or gonioscopic examination
Slit-lamp or gonioscopic examination
 Serum copper and ceruloplasmin levels, urine copper level
Serum copper and ceruloplasmin levels, urine copper level
Treatment
 Treatment by an internist and/or neurologist with copper chelating agents such as D-penicillamine or tetrathiomolybdate
Treatment by an internist and/or neurologist with copper chelating agents such as D-penicillamine or tetrathiomolybdate
Prognosis
 Good with early recognition and treatment. The Kayser-Fleischer ring may resolve with treatment.
Good with early recognition and treatment. The Kayser-Fleischer ring may resolve with treatment.
FIGURE 8-1. Wilson’s disease. A prominent Kayser-Fleischer ring can be seen. Brown copper pigment deposition is very apparent in the corneal periphery in this 18-year-old woman with Wilson’s disease. The deposits are at the level of Descemet’s membrane and deep stroma. In mild cases, the copper pigment is seen earliest using gonioscopy. See also Chapter 6, Figure 6-8.
VITAMIN A DEFICIENCY
Vitamin A deficiency is a rare, potentially blinding disorder that usually affects the malnourished and is common in areas where polished rice is the main source of food.
Etiology
 Dietary deficiency of vitamin A, typically from chronic malnutrition
Dietary deficiency of vitamin A, typically from chronic malnutrition
 Celiac diseases, biliary obstruction, cystic fibrosis, pancreatic or intestinal (e.g., gastric band or stapling) surgery, which impairs absorption of vitamin A
Celiac diseases, biliary obstruction, cystic fibrosis, pancreatic or intestinal (e.g., gastric band or stapling) surgery, which impairs absorption of vitamin A
Symptoms
 Night blindness is the earliest symptom; dry eye, foreign-body sensation, gradual loss of vision in severe cases
Night blindness is the earliest symptom; dry eye, foreign-body sensation, gradual loss of vision in severe cases
Signs
 Xerosis (severe drying) of cornea and conjunctiva
Xerosis (severe drying) of cornea and conjunctiva
 Keratinization of conjunctiva (Bitot’s spot: superficial, triangular, silvery-gray, foamy, keratinized patch on the interpalpebral bulbar conjunctiva)
Keratinization of conjunctiva (Bitot’s spot: superficial, triangular, silvery-gray, foamy, keratinized patch on the interpalpebral bulbar conjunctiva)
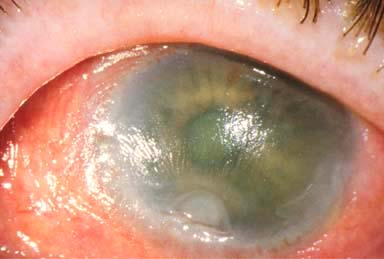
 Sterile corneal ulcers and melts (keratomalacia), which may lead to scarring or perforation (Fig. 8-2)
Sterile corneal ulcers and melts (keratomalacia), which may lead to scarring or perforation (Fig. 8-2)
Differential Diagnosis
 Keratoconjunctivitis sicca
Keratoconjunctivitis sicca
Diagnostic Evaluation
 Serum vitamin A level
Serum vitamin A level
 Consider impression cytology of the conjunctiva (demonstrates decreased goblet cell density) and electroretinogram.
Consider impression cytology of the conjunctiva (demonstrates decreased goblet cell density) and electroretinogram.
Treatment
 Systemic vitamin A orally or intramuscularly and repeated 1 month later
Systemic vitamin A orally or intramuscularly and repeated 1 month later
 Frequent preservative-free artificial tear drops or ointment to lubricate ocular surface
Frequent preservative-free artificial tear drops or ointment to lubricate ocular surface
 Treat of the malnutrition
Treat of the malnutrition
 Corneal transplantation for scars or perforation. Consider a keratoprosthesis for bilateral severe scarring with good macular function.
Corneal transplantation for scars or perforation. Consider a keratoprosthesis for bilateral severe scarring with good macular function.
Prognosis
 Very good if diagnosed and treated before significant corneal ulceration has occurred. Fair to poor if significant corneal damage is present.
Very good if diagnosed and treated before significant corneal ulceration has occurred. Fair to poor if significant corneal damage is present.
FIGURE 8-2.Vitamin A deficiency. This malnourished patient had severe xerosis of the cornea and conjunctiva. There was a deep sterile corneal melt near the limbus from the 6 to the 7 o’clock position. The xerosis and melt resolved over a week with systemic vitamin A therapy.
CYSTINOSIS
Cystinosis is a rare disorder that results in accumulation of cystine in the body.
Etiology and Epidemiology
 Autosomal recessive disorder
Autosomal recessive disorder
 Results in deposits of the amino acid cystine in the conjunctiva, corneal stroma, iris, lens, and retina, depending on severity
Results in deposits of the amino acid cystine in the conjunctiva, corneal stroma, iris, lens, and retina, depending on severity
 Three main forms
Three main forms
 Infantile: associated with dwarfism and progressive kidney dysfunction. Poor prognosis without a renal transplant
Infantile: associated with dwarfism and progressive kidney dysfunction. Poor prognosis without a renal transplant
 Adolescent (intermediate): Kidneys may be involved, but retinas are normal.
Adolescent (intermediate): Kidneys may be involved, but retinas are normal.
 Adult: minimal to no kidney disease, cystine deposits limited to anterior segment
Adult: minimal to no kidney disease, cystine deposits limited to anterior segment
Symptoms
 Irritation, foreign-body sensation, pain, occasionally decreased vision
Irritation, foreign-body sensation, pain, occasionally decreased vision
Signs
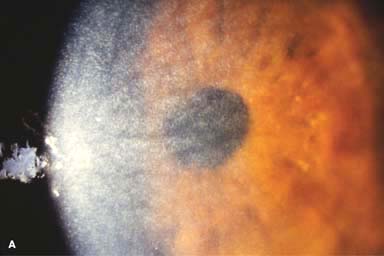
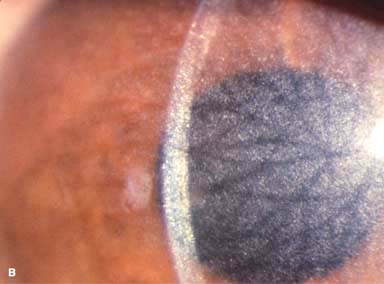
 Myriad tiny, glistening crystals in the corneal stroma, conjunctiva, iris, lens, and retina, depending on the severity of the disease (Fig. 8-3)
Myriad tiny, glistening crystals in the corneal stroma, conjunctiva, iris, lens, and retina, depending on the severity of the disease (Fig. 8-3)
 May have superficial punctate keratopathy, filaments, and recurrent erosions
May have superficial punctate keratopathy, filaments, and recurrent erosions
 Growth retardation, renal failure, hepatosplenomegaly, and hypothyroidism
Growth retardation, renal failure, hepatosplenomegaly, and hypothyroidism
Differential Diagnosis
 See Crystalline Keratopathy in Chapter 6.
See Crystalline Keratopathy in Chapter 6.
Treatment
 Lubrication for ocular surface disease
Lubrication for ocular surface disease
 Cysteamine eye drops have been reported to be useful.
Cysteamine eye drops have been reported to be useful.
 Rarely, a corneal transplant is required.
Rarely, a corneal transplant is required.
Prognosis
 Poor for the infantile form; good for the intermediate and adult forms.
Poor for the infantile form; good for the intermediate and adult forms.
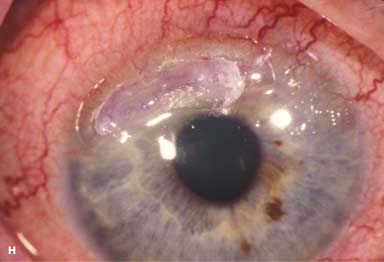
FIGURE 8-3.Cystinosis.A. Confluent full-thickness tiny refractile corneal deposits are seen. These opacities are cystine crystals. Generally, the deposits do not affect vision; however, if they are severe, they can cause significant visual symptoms. B. Slit-beam view of the eye with cystinosis seen in A. Note the full-thickness distribution of the crystals.
MUCOPOLYSACCHARIDOSES AND LIPIDOSES
The mucopolysaccharidoses and lipidoses comprise a group of inherited systemic metabolic disorders that result in abnormal accumulation of material in the body.
Etiology and Epidemiology
 Mucopolysaccharidoses: lysosomal storage diseases, including Hurler’s, Scheie’s, Hunter’s, Sanfilippo’s, Morquio’s, Maroteaux-Lamy, and Sly’s syndromes. All are autosomal recessive except Hunter’s syndrome, which is X-linked recessive.
Mucopolysaccharidoses: lysosomal storage diseases, including Hurler’s, Scheie’s, Hunter’s, Sanfilippo’s, Morquio’s, Maroteaux-Lamy, and Sly’s syndromes. All are autosomal recessive except Hunter’s syndrome, which is X-linked recessive.
 Lipidoses: Numerous disorders of lipid metabolism, including Fabry’s disease. All lipidoses are autosomal recessive except Fabry’s disease, which is X-linked recessive.
Lipidoses: Numerous disorders of lipid metabolism, including Fabry’s disease. All lipidoses are autosomal recessive except Fabry’s disease, which is X-linked recessive.
Signs
 Mucopolysaccharidoses: All may have optic nerve, retinal, and CNS abnormalities. All have progressive corneal clouding except Hunter’s and Sanfilippo’s syndromes (Fig. 8-4).
Mucopolysaccharidoses: All may have optic nerve, retinal, and CNS abnormalities. All have progressive corneal clouding except Hunter’s and Sanfilippo’s syndromes (Fig. 8-4).
 Lipidoses: All may have macular cherry-red spots and optic atrophy. Bilateral, symmetric, brownish corneal epithelial deposits arranged in a vortex fashion from a point below the pupil and swirling outward but sparing the limbus (cornea verticillata) are seen in males with Fabry’s disease and female carriers. Conjunctival aneurysms, lens opacities, papilledema, optic atrophy, and macular and retinal edema are also seen in Fabry’s disease.
Lipidoses: All may have macular cherry-red spots and optic atrophy. Bilateral, symmetric, brownish corneal epithelial deposits arranged in a vortex fashion from a point below the pupil and swirling outward but sparing the limbus (cornea verticillata) are seen in males with Fabry’s disease and female carriers. Conjunctival aneurysms, lens opacities, papilledema, optic atrophy, and macular and retinal edema are also seen in Fabry’s disease.
Treatment
 Severe corneal opacity may require a corneal transplant. No ocular treatment is required for cornea verticillata.
Severe corneal opacity may require a corneal transplant. No ocular treatment is required for cornea verticillata.
 Follow-up with a pediatrician or pediatric endocrinologist and a geneticist
Follow-up with a pediatrician or pediatric endocrinologist and a geneticist
Prognosis
 Poor to good, depending on the specific metabolic disorder
Poor to good, depending on the specific metabolic disorder
FIGURE 8-4.Maroteaux-Lamy syndrome. Diffuse full-thickness corneal haze can be seen in this child with Maroteaux-Lamy syndrome.
COLLAGEN VASCULAR DISEASES
Collagen vascular diseases can cause a wide variety of ocular abnormalities, the most important of which are peripheral ulcerative keratitis and scleritis.
Etiology
 Rheumatoid arthritis (most common)
Rheumatoid arthritis (most common)
 Wegener’s granulomatosis (often causes a necrotizing scleritis)
Wegener’s granulomatosis (often causes a necrotizing scleritis)
 Polyarteritis nodosa
Polyarteritis nodosa
 Relapsing polychondritis
Relapsing polychondritis
 Systemic lupus erythematosus (SLE)
Systemic lupus erythematosus (SLE)
 Others
Others
Symptoms
 Range from none to significant pain, redness, discharge, and decreased vision
Range from none to significant pain, redness, discharge, and decreased vision
Signs
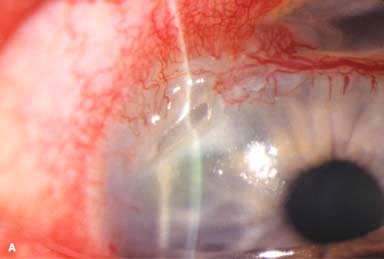
 Corneal findings include keratoconjunctivitis sicca, stromal keratitis, corneal stromal infiltrates or ulceration, which is typically peripheral but may be central. The peripheral corneal ulceration may occur with or without symptomatic inflammation. The ulceration can be similar to Mooren’s ulcer in that it can extend circumferentially and centrally. However, unlike Mooren’s ulcer, the sclera is commonly involved. Corneal perforation may occur (Fig. 8-5A–C).
Corneal findings include keratoconjunctivitis sicca, stromal keratitis, corneal stromal infiltrates or ulceration, which is typically peripheral but may be central. The peripheral corneal ulceration may occur with or without symptomatic inflammation. The ulceration can be similar to Mooren’s ulcer in that it can extend circumferentially and centrally. However, unlike Mooren’s ulcer, the sclera is commonly involved. Corneal perforation may occur (Fig. 8-5A–C).
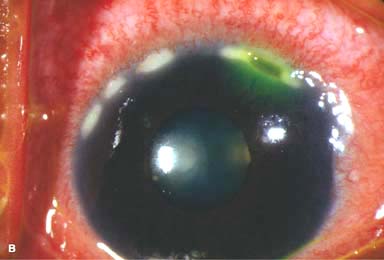
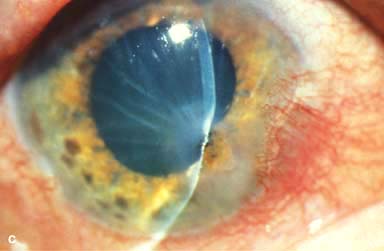
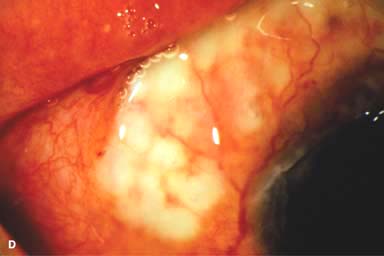
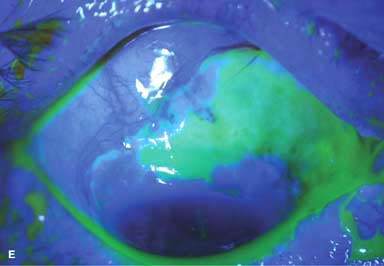
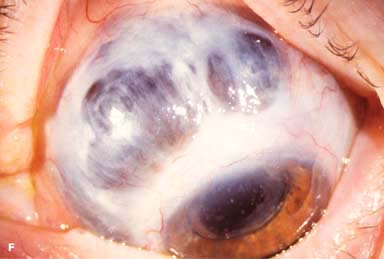
 Other findings include episcleritis, scleritis (necrotizing with or without inflammation) and sclerokeratitis (Fig. 8-5D and E). Healed episodes of scleritis can lead to scleral thinning and uveal show (Fig. 8-5F).
Other findings include episcleritis, scleritis (necrotizing with or without inflammation) and sclerokeratitis (Fig. 8-5D and E). Healed episodes of scleritis can lead to scleral thinning and uveal show (Fig. 8-5F).
 The corneal changes in SLE are often unremarkable.
The corneal changes in SLE are often unremarkable.
Differential Diagnosis
 Mooren’s ulcer: no underlying systemic disease
Mooren’s ulcer: no underlying systemic disease
 Infectious infiltrate or ulcer: pain, iritis, more purulent discharge, cultures positive
Infectious infiltrate or ulcer: pain, iritis, more purulent discharge, cultures positive
Treatment
 Artificial tear drops, gels, and ointment, cyclosporine 0.05% drops and punctal occlusion are used for keratoconjunctivitis sicca. Topical corticosteroids are helpful in stromal keratitis but should generally be avoided in corneal and scleral ulcers. Topical cyclosporine 0.5% to 2% q.i.d. may also be beneficial.
Artificial tear drops, gels, and ointment, cyclosporine 0.05% drops and punctal occlusion are used for keratoconjunctivitis sicca. Topical corticosteroids are helpful in stromal keratitis but should generally be avoided in corneal and scleral ulcers. Topical cyclosporine 0.5% to 2% q.i.d. may also be beneficial.
 Oral nonsteroidal anti-inflammatory agents and/or corticosteroids are used for peripheral ulcerative keratitis and scleritis. Topical treatment is aimed at reepithelialization and prevention of secondary infection (e.g., artificial tear and antibiotic ointment q.i.d., punctal occlusion, lateral tarsorrhaphy).
Oral nonsteroidal anti-inflammatory agents and/or corticosteroids are used for peripheral ulcerative keratitis and scleritis. Topical treatment is aimed at reepithelialization and prevention of secondary infection (e.g., artificial tear and antibiotic ointment q.i.d., punctal occlusion, lateral tarsorrhaphy).
 Stronger immunosuppressive agents (e.g., methotrexate, azathioprine, cyclosporine, or cyclophosphamide) or biologics (e.g., infliximab, adalimumab, or rituximab) may be required, especially for scleritis or severe corneal stromal inflammation.
Stronger immunosuppressive agents (e.g., methotrexate, azathioprine, cyclosporine, or cyclophosphamide) or biologics (e.g., infliximab, adalimumab, or rituximab) may be required, especially for scleritis or severe corneal stromal inflammation.
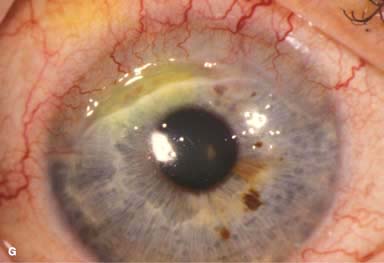
 Resection of inflamed conjunctiva adjacent to a peripheral corneal ulcer may be helpful on rare occasions. Cyanoacrylate tissue glue can be used for small perforations (Fig. 8-5G and H). Larger perforations will require corneal patch grafts.
Resection of inflamed conjunctiva adjacent to a peripheral corneal ulcer may be helpful on rare occasions. Cyanoacrylate tissue glue can be used for small perforations (Fig. 8-5G and H). Larger perforations will require corneal patch grafts.
Prognosis
 Fair to good, depending on the severity and response of the underlying systemic disease
Fair to good, depending on the severity and response of the underlying systemic disease
FIGURE 8-5. Rheumatoid arthritis.A. Slit-beam view of a patient with rheumatoid arthritis demonstrates a peripheral corneal melt with severe ulceration. There is approximately 90% tissue loss. There is moderate corneal neovascularization peripherally and superiorly. Note the area of clear cornea superiorly; it is an additional large area of corneal melting. B. This patient with rheumatoid arthritis has three separate peripheral corneal infiltrates from the 9 to the 11 o’clock positions. There is an additional, larger peripheral infiltrate with corneal melting from the 12 to the 2 o’clock positions. The infiltrates are most likely sterile, inflammatory infiltrates. Rheumatoid arthritis.C. This eye has a large sterile corneal melt leading to perforation in the midperiphery at the 4:30 position. The radiating stromal folds and peaked pupil suggest a perforation. Peripheral corneal neovascularization and scarring from the 3 to the 4 o’clock positions indicate 0 previous corneal inflammation in that area. Wegener’s granulomatosis.D. This patient with a necrotizing scleritis and peripheral corneal melt had no known medical problems. Emergency systemic work-up revealed Wegener’s granulomatosis. She was treated aggressively with systemic corticosteroids and cyclophosphamide, and her scleritis resolved. Appropriate diagnosis and treatment of Wegener’s granulomatosis can be life-saving. Wegener’s granulomatosis. E. A large fluorescein-staining conjunctival epithelial defect overlying a necrotizing scleritis can be seen in this patient with Wegener’s granulomatosis. Rheumatoid arthritis.F. This eye had previous, asymptomatic scleral inflammation (scleritis) with progressive loss of scleral tissue. The sclera has become so thin that the brown uveal tissue can be seen. This condition is termed scleromalacia perforans. Rheumatoid arthritis. G. A large crescentic area of peripheral ulcerative keratitis can be seen in this patient with rheumatoid arthritis. The anterior chamber was flat, and a small perforation was found in the ulceration at the 10 o’clock position. H. Due to the perforation and flat anterior chamber, the eye was treated with cyanoacrylate tissue glue. The anterior chamber re-formed nicely over the next hour.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


