Fig. 9.1
Eyelid fibromatosis. A 6-month-old child with a tethering of the eyelid to a deeper firm mass in the superonasal aspect with consequent lagophthalmos (a). Histopathology examination shows infiltrating bundles of spindle cells with lobulated pattern in few areas diagnostic of fibromatosis (b, hematoxylin and eosin, original magnification ×200)
9.1.5 Fibrosarcoma
Fibrosarcoma is a highly malignant tumor that can be locally destructive and can metastasize.
9.1.5.1 Clinical Features
9.1.5.2 Histopathologic Features
The lesion consists of closely packed cells that assume an interlacing woven herringbone pattern. The cells contain a vesicular nucleus with prominent nucleoli, tapering pointed ends with moderate mitotic activity.
9.2 Fibrohistiocytic Tumors
Fibrohistiocytic tumors could be subclassified as benign (xanthelasma, xanthoma, dermatofibroma, xanthogranuloma, juvenile xanthogranuloma, reticulohistiocytoma), intermediate (atypical fibroxanthoma, dermatofibrosarcoma protuberans, angiomatoid fibrous histiocytoma), and malignant (malignant fibrous histiocytoma, malignant fibroxanthoma).
9.2.1 Xanthelasma
Xanthelasma palpebrarum is a common bilateral subcutaneous lesion of the eyelid seen in normolipemic individuals and in those with primary hyperlipemia (types II and III) or secondary hyperlipemia (diabetes mellitus, biliary cirrhosis) [7]. It was found that alteration in apolipoprotein levels in patients with xanthelasma may predispose to cutaneous and systemic depositions of lipids, including atheroscleosis [8].
9.2.1.1 Clinical Features
Xanthelasma manifests as a yellowish-tan soft plaque occurs in the inner canthus in middle-aged individuals (Fig. 9.2). Large nodular xanthelasma is called xanthoma or tuberous xanthoma, which has a known association with Erdheim–Chester disease. Acute-onset eruptive xanthoma occurs in patients experiencing a rapid rise in serum triglyceride levels. Management should include systemic evaluation for the causative etiology and excision or laser [9] or radiofrequency-assisted vaporization of large cosmetically unacceptable lesions [10].
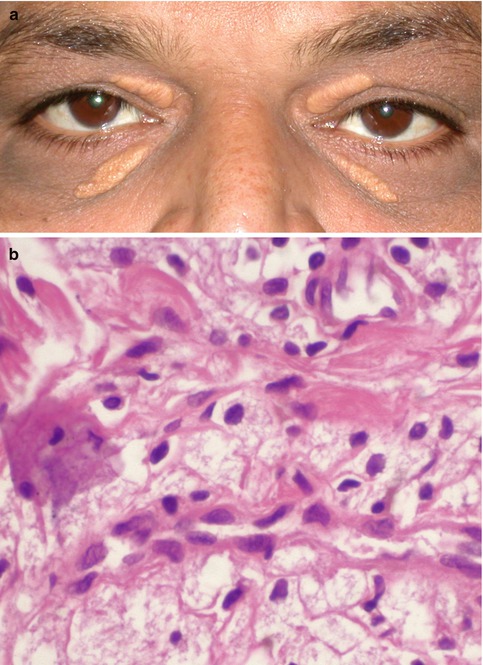
Fig. 9.2
Xanthelasma. Bilateral yellowish placoid lesions clinically diagnostic of xanthelasma (a). Sheets of large, foamy lipid-laden cells on histopathology (b, hematoxylin and eosin, original magnification × 400)
9.2.1.2 Histopathologic Features
Microscopically, xanthelasma consists of lipid-laden macrophages in the superficial dermis, around blood vessels, and adnexa.
9.2.2 Xanthogranuloma
9.2.2.1 Clinical Features
Juvenile xanthogranuloma of the eyelid may be a part of systemic affection seen as multiple fleshy superficial eyelid nodules with or without coexisting conjunctival, iris, and orbital involvement. The adult variant may be diffuse and associated with bronchial asthma (Fig. 9.3) [11]. Juvenile xanthogranuloma is known to spontaneously involute, thus qualifying for observation. Systemic corticosteroids are indicated in recalcitrant juvenile xanthogranuloma and as primary therapy for the adult variant [11]. Extensive, cosmetically disfiguring, and steroid-resistant lesions could be excised or treated with systemic chemotherapy or radiotherapy.
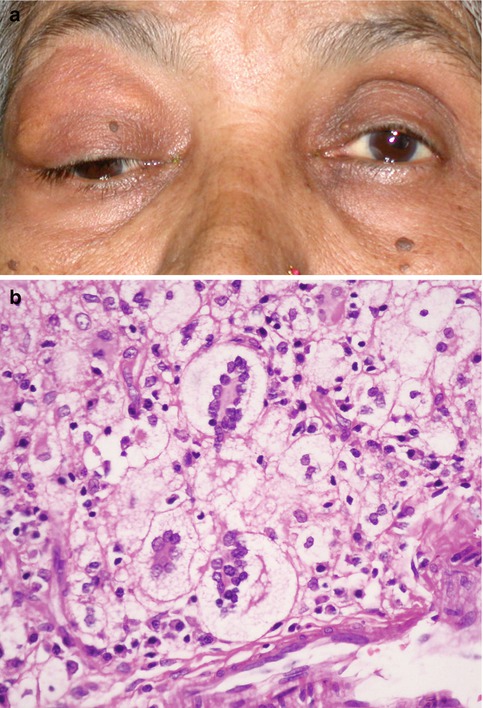
Fig. 9.3
Adult-onset xanthogranuloma presenting as a diffuse eyelid mass with a yellowish haze (a). Sheets of foamy histiocytes and classic Touton type of multinucleated giant cells with wreath-like arrangement of the nuclei on histopathology (b, hematoxylin and eosin, original magnification ×250)
9.2.2.2 Histopathologic Features
Xanthogranuloma consists of monomorphic infiltrate of histiocytes, intermixed with lymphocytes and the classic Touton giant cell with a wreath-like arrangement of nuclei and peripheral clear zone.
9.2.3 Malignant Fibrous Histiocytoma
Malignant fibrous histiocytoma is a pleomorphic soft tissue sarcoma that occurs rarely in the eyelid.
9.2.3.1 Clinical Features
Malignant fibrous histiocytoma presents as a firm subcutaneous mass [13]. Dermatofibrosarcoma protuberans, which manifests as a rapidly growing eyelid nodule, is considered an aggressive form of malignant fibrous histiocytoma [14]. It has tendency for local invasion and is known to metastasize. The mainstay of treatment is complete surgical excision with wide margins. Because of risk of recurrence following excision, consideration should be given to histologic margin control and adjuvant radiotherapy [15].
9.2.3.2 Histopathologic Features
Malignant fibrous histiocytoma differs from benign variant in exhibiting marked nuclear pleomorphism, high mitotic activity, pericytoma-like areas with foci of xanthoma cells, and multinucleated giant cells.
9.3 Lipomatous Tumors
Lipoma, lipoma variants, and liposarcoma are the lipomatous tumors affecting the eyelid [16].
9.3.1 Lipoma and Lipoma Variants
Lipoma could be congenital or acquired. Nasopalpebral lipoma–coloboma syndrome is an autosomal dominant syndrome characterized by congenital upper eyelid and nasopalpebral lipomas, upper and lower eyelid colobomas, telecanthus, and maxillary hypoplasia [17]. Lipoblastoma is an uncommon benign tumor of adipose tissue that occurs in infants and young children [18]. Intramuscular lipoma of the eyelid manifesting as a slowly growing globular lesion has been described in elderly individuals. Hibernoma is a variant of lipoma that contains embryonal brown fat and is relatively more vascular. If treatment is indicated for cosmetic and functional reasons, complete tumor excision is adequate.
9.3.2 Liposarcoma
Primary liposarcoma is a rare orbital tumor that may involve the eyelid by local extension.
9.4 Smooth Muscle Tumors
9.5 Skeletal Muscle Tumors
Rhabdomyoma and rhabdomyosarcoma are the skeletal muscle tumors of the eyelid.
9.5.1 Rhabdomyoma
Rhabdomyoma is a benign tumor of the skeletal muscle and is seen in two forms. The adult form consists of well-differentiated large rounded or polygonal cells with abundant acidophilic cytoplasm containing lipid and glycogen. Some cells appear like spider cells and some may show cross striations. The fetal form is very cellular and consists of immature skeletal muscle fibers and primitive mesenchymal cells. One case of adult-onset rhabdomyoma attributed to chronic irritation by a prosthetic eye has been reported in the literature [21].
9.5.2 Rhabdomyosarcoma
Rhabdomyosarcoma is primarily a malignant orbital tumor with eyelid involvement only in about 3 % of cases [22].
9.6 Vascular Tumors
Benign vascular tumors of the eyelid include nevus flammeus, papillary endothelial hyperplasia, capillary hemangioma, cavernous hemangioma, venous hemangioma, epithelioid hemangioma, arteriovenous malformation, and lymphangioma. Angiosarcoma, lymphangiosarcoma, and Kaposi’s sarcoma are the malignant vascular eyelid tumors.
9.6.1 Nevus Flammeus (Port-Wine Stain)
Nevus flammeus is a diffuse congenital vascular malformation of the face, involving the periocular area and eyelid (Chap. 12).
9.6.2 Pyogenic Granuloma
Papillary endothelial hyperplasia or “pyogenic granuloma” is the most common acquired vascular lesion of the eyelid. It is neither “pyogenic” nor is it a “granuloma.”
9.6.2.1 Clinical Features
Pyogenic granuloma occurs anywhere in the eyelid, as a rapidly growing, pedunculated reddish-pink mass with or without superficial ulceration and may easily bleed on touch (Fig. 9.4). It usually follows trauma or surgery. Local excision of the lesion is curative [23].
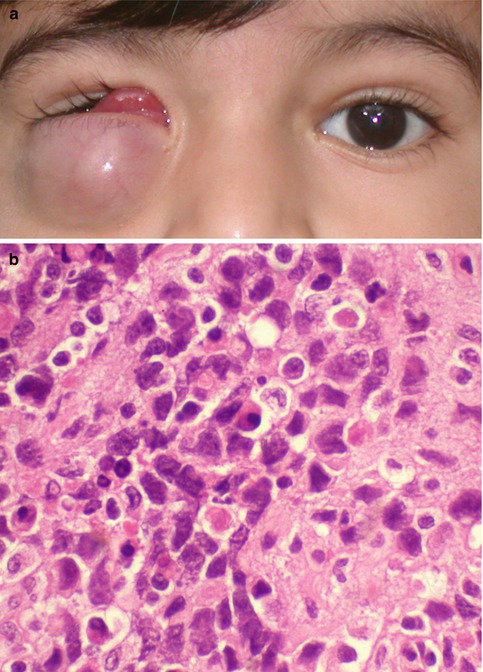
Fig. 9.4
Pyogenic granuloma. The tarsal conjunctiva of the upper eyelid showing a vascular polypoidal reddish-pink mass with superficial ulceration (a). Histopathologically, loose edematous stroma with surface necrosis, proliferating blood vessels, and mixed inflammatory infiltrates, characteristic of inflammatory granulation tissue, is present (b, hematoxylin and eosin, original magnification ×200)
9.6.2.2 Histopathologic Features
Pyogenic granuloma consists of exuberant mass of proliferating radiating capillaries and edematous stroma with mixed inflammatory infiltrates. Intravascular papillary endothelial hyperplasia is a rare form of “pyogenic granuloma” in which the angiomatous proliferation is confined entirely within the lumen of a vessel [24].
9.6.3 Capillary (Infantile) Hemangioma
Capillary hemangioma of the eyelid is the most common vascular tumor of the eyelid in children (Fig. 9.5). It is usually congenital in origin and is often sporadic. Newborns of mothers who have undergone amniocentesis and in premature infants are at a risk of developing capillary hemangioma [25]. The pathogenesis of this tumor is not well understood, but the affected infants have an increased urinary level of basic fibroblastic growth factor, a marker of angiogenesis. Familial congenital capillary hemangioma with autosomal dominant inheritance with incrimination of chromosome 5q has been reported [26].
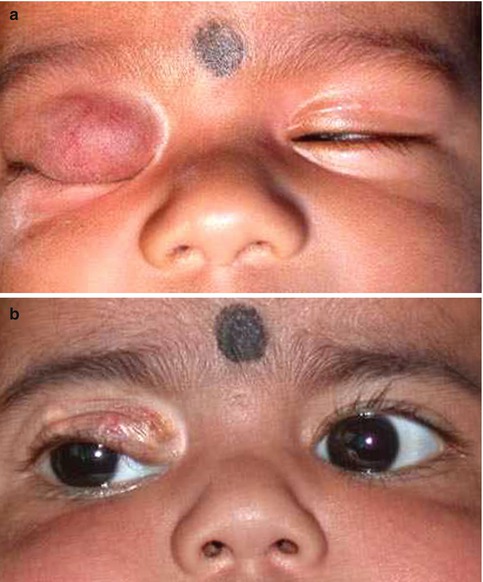
Fig. 9.5
Capillary hemangioma of the upper eyelid manifesting as a bright red, spongy, soft mass causing total ptosis (a). It resolved following treatment with intralesional triamcinolone injection (b)
9.6.3.1 Clinical Features
Congenital capillary hemangioma usually manifests at birth or within the first months of life. There are two distinct clinical variants – superficial and deep. The superficial variant, better known as strawberry hemangioma, appears as a bright red to deep purple, lobulated, spongy, soft eyelid mass that typically blanches on application of direct pressure and engorges when the infant cries or strains. Contiguous conjunctival and orbital extension is known to occur. The superficial variant is localized to the epidermis and dermis, whereas the deep variant lies in the subcutaneous tissue and is bluish or blue gray in color.
9.6.3.2 Natural History
9.6.3.3 Histopathologic Features
Histologically, capillary hemangioma of the eyelid consists of lobules of capillaries separated by sparse fibrous septae. The morphology of the lesion changes with age. An early immature lesion tends to have obliterated lumen with plump endothelial cells and occasional mitotic figures (Fig. 9.6), while in later stages the lumina increases and the endothelial cells get attenuated, with increasing fibrosis and fat infiltration.
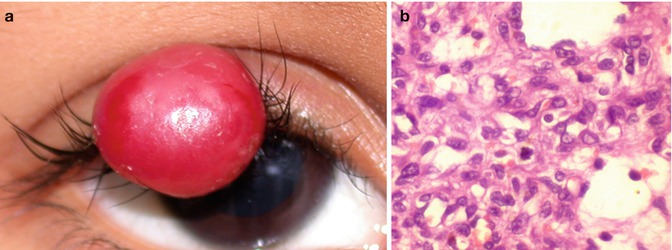
Fig. 9.6
An older child with a large red vascular mass in the upper eyelid (a) that was excised. Histopathology shows lobulated appearance with vascular channels lined by plump endothelial cells. Note the presence of a few mitotic figures could be seen in proliferating lesions (b, hematoxylin and eosin, original magnification ×400)
9.6.3.4 Systemic Association
In most instances, congenital capillary hemangioma is a sporadic condition but approximately 20 % of patients may manifest it as multiple tumors involving the cutaneous tissue elsewhere, central nervous system, liver, and gastrointestinal tract [25]. Systemic lesions, especially those found in association with Kasabach–Merritt syndrome, could aggressively proliferate and lead to hemorrhage, platelet consumption, disseminated intravascular coagulation, cardiac failure, and death [25].
9.6.3.5 Treatment
The main ocular complications are amblyopia and strabismus. Amblyopia could be meridional because of induced astigmatism or because of stimulation deprivation secondary to mechanical ptosis. Because most lesions spontaneously regress, observation, refractive correction, and appropriate amblyopia management are the standard treatment. Active intervention is indicated if the lesion extensively involves the face or is ulcerated with episodes of bleeding and if there is mechanical ptosis with obscuration of pupillary axis or induced astigmatism with amblyopia. Extensive lesions are treated with oral prednisolone 1–2 mg/kg body weight tapered over 4–6 weeks. Application of topical clobetasol propionate may help [28]. Intralesional steroid injection (Fig. 9.5) is mainly reserved for eyelid and anterior orbital lesions. Most lesions regress after 1–3 injections of triamcinolone or a combination of dexamethasone and triamcinolone injected at 6–8 weekly intervals [29]. The recommended dosage per injection is 6 mg/kg body weight equivalent of prednisolone. Although uncommon, reported complications of intralesional steroid injection include central retinal artery occlusion, eyelid depigmentation, fat atrophy, eyelid necrosis, and adrenal suppression [29]. Alternative treatment modalities include interferon, laser sclerotherapy, and excision of circumscribed anterior lesions (Fig. 9.6).
In recent years several centers have reported the use of systemic β-blockers for the treatment of infantile hemangioma, with very promising results (Fig. 9.7) [30–32]. The dose that has been used is 0.5–2.0 mg/kg/day for several months, with significant reduction in the size of the hemangioma in all babies. Side effects such as bradycardia, hypotension, bronchospasm, hypoglycemia, and electrolyte disturbances have been reported, leading to a reduction in the dose of the drug. Topical timolol maleate 0.5 % was also found to be effective [33].
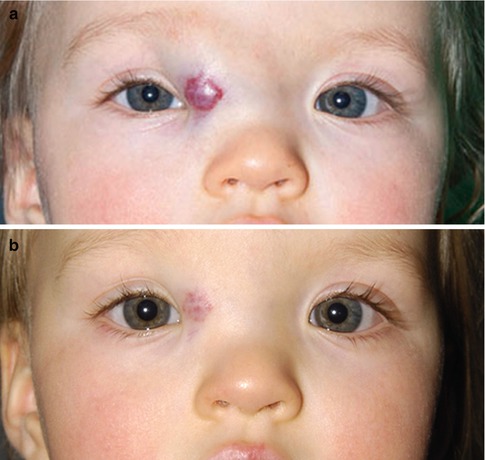
Fig. 9.7
Capillary hemangioma. A 10-month-old female with history of failed intralesional steroid injection. After pediatric cardiology examination, BP, EKG, and echocardiogram, propranolol was started at 1 mg/kg/day in three divided doses daily (a, 11/11/2010), with a blood glucose level and repeat blood pressure check after the first dose (blood pressure, taken in the left thigh 87/61; pulse 108; glucose 80; about 2 h after the first increased dose). One week later (11/18/2010) the dosage was doubled to 2 mg/kg/day in three divided doses daily. BP, heart rate, and blood glucose were again checked about 2 h after the first increased dose. No additional testing was performed for increases above 2 mg/kg/day. Her dose was increased to 3 mg/kg/day (12/9/2010). After 3 months of treatment, the infantile hemangioma had shown marked regression (b, 2/10/11). The dose was then tapered to 1.5 mg/kg/day (4/13/2011), 1 mg/kg/day (5/25/2011), 0.5 mg/kg/day (6/15/2011), and finally discontinued on 7/1/2011. No regrowth of infantile hemangioma was observed (Courtesy of Paul Rychwalski, MD and Alex Golden, MD, Cleveland Clinic, Ohio)
9.6.4 Cavernous Hemangioma
Cavernous hemangioma of the eyelid is a rare, acquired condition and is generally seen in adults [34]. It may be associated with blue rubber bleb nevus syndrome [35].
9.6.4.1 Clinical Features
The lesions are ill circumscribed and bluish in color and may undergo slow progression. Epithelioid hemangioma also known as angiolymphoid hyperplasia with eosinophilia occurs as a nodular lesion in the eyelid. Kimura disease, which is predominant in Asian population, shares both clinical and histopathologic features with angiolymphoid hyperplasia with eosinophilia and may be clinically indistinguishable [36].
9.6.4.2 Histopathologic Features
Cavernous hemangioma is composed of large dilated vascular channels filled with blood, hemosiderin-laden macrophages, scattered lymphoplasmacytic infiltrates, and secondary changes such as calcification, phlebolith, and fibrosis.
9.6.5 Arteriovenous Malformations
Arteriovenous malformations, as the name suggests, are communications between arteries and veins that bypass normal capillary beds. In contrast to arteriovenous fistulas, arteriovenous malformations are mainly congenital lesions with multiple large feeding arteries, a central nidus, and numerous dilated draining veins (Fig. 9.8). Rarely, arteriovenous malformations may follow trauma or surgery [37]. While surgical embolization or excision alone may be possible, a combined approach is considered ideal [37].
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
