Fig. 7.1
Macular degeneration is caused by degeneration of the retina resulting in vision loss in the macula at the center of the visual field. As this simulated photographic representation shows, macular degeneration makes it difficult to focus on central objects and recognize faces. However, the condition leaves peripheral vision intact
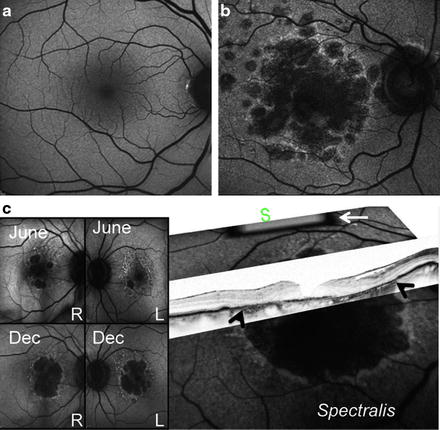
Fig. 7.2
Retinal cell loss in macular degeneration. (a) Normal fundi from a healthy subject. (b) Fundi from a patient with advanced macular degeneration showing RPE loss. (c) A2E autofluorescence (AF) images (over 18 months, top to bottom) of non-exudate age-related macular degeneration, showing progressive RPE loss. This patient is double-homozygous for ARMS2 (T-in/del), HTRA1 (a), and CFH (402Y) and non-cGMP grade iPS has already been generated. In 2007, visual acuity was 20/40 in right (RE) and 20/25 left eye (LE). In 2011, her vision was 20/400 (RE) and 20/400 and the patient lost her independence in activities of daily living. Scattered, nonconfluent drusen are visible at the posterior pole, along with minor pigmentary alterations. Expanding spots of RPE loss can be seen in the area of increased AF nasal and superior to the large central spot of atrophy. A higher AF signal indicates excessive amounts of lipofuscin in the retinal sites that will continue to undergo RPE death, leading to absolute scotoma (areas of vision loss). White arrow indicates the internal fluorescence reference rectangle, which is mounted in the intermediate retinal plane of the camera. The reference is in focus with the image and can account for variable laser power and detector sensitivity during image analysis. White arrows on optical coherence tomography (OCT) mark the loss of RPE. S, superior macular region
Although AMD is not a hereditary macular dystrophy, several susceptibility genes have been identified [2–5]. Complement factor H (CHF Y402H haplotype), which inhibits the complement cascade from reacting against the body’s own tissues, is a major susceptibility gene in AMD [2]. The SERPING1 gene product and polymorphisms of the SERPING1 gene, which regulate the classical complement pathway, are also associated with AMD [3]. Genes associated with oxidative stress have also been implicated. The LOC387715/ARMS2 gene on chromosome 10q26, which produces a protein found in the mitochondrial outer membrane, is the second major susceptibility gene identified in AMD [4, 5]. Together, CHF Y402H, SERPING1, and LOC387715/ARMS2 are found in over 60 % of cases of AMD. Although genome-wide association studies have identified a number of genes, including those described above, that are highly associated with AMD, the underlying molecular mechanisms that ultimately lead to RPE loss are not known. The mechanisms that lead to the cells’ disappearance are likely related to a mixture of both hereditary and environmental factors.
The photoreceptor/RPE complex is also preferentially affected in Stargardt disease, the most prevalent of the hereditary macular dystrophies. Stargardt disease leads to a progressive loss of central visual acuity in the first two decades of life. ABCA 4 is a gene found to be mutated in 70 % of Stargardt cases [6]. ABCA4 encodes for a protein called Rim, located in the rims of photoreceptor disc membranes, which is involved in the transportation of a vitamin A intermediate to the RPE, where it prevents lipofuscin from accumulating [7]. Lipofuscin accumulation results from the incomplete digestion of phagocytosed outer segment photoreceptors in the RPE. A major component of RPE lipofuscin is A2E, a by-product of the visual cycle [8]. ABCA4 mice, an animal model for Stargardt disease, develop an accelerated accumulation of A2E in the RPE, thickening of the Bruch membrane, and visual loss [9]. Clinically, Stargardt disease results in lipofuscin accumulation in the RPE and photoreceptor inner segments, RPE and choroidal vascular atrophy, macular photoreceptor loss, and reactive Müller glial hypertrophy. A retrovirus carrying the wild-type ABCA4 gene was found to reduce lipofuscin accumulation in a mouse model of Stargardt disease [10].
Retinitis pigmentosa (RP) is a heterogeneous group of disorders characterized by a progressive deterioration of rod and cone photoreceptor function (see Fig. 7.3). Currently, nearly 250 different genes and mapped loci have been found to be implicated in RP, which is the leading cause of inherited blindness in children and young adults. One form of retinitis pigmentosa, Leber congenital amaurosis, is a rare but severe inherited rod-cone dystrophy affecting children within the first year of life. More than a dozen genes have been implicated in the condition. For instance, RPE65, implicated in Leber congenital amaurosis type 2, codes for a protein found in the RPE that recycles the by-product of the visual cycle all-trans-retinal back to 11-cis-retinal. Gene therapy introducing RPE65 has restored retinal function to Briard dogs that have a mutated version of the RPE65 gene [11]. Human trials have attempted to restore sight in Leber congental amaurosis using the same viral vectors to introduce the RPE65 gene to study subjects, but have demonstrated mixed results [12].

Fig. 7.3
In contrast with macular degeneration, retinitis pigmentosa causes narrowing of peripheral vision until the visual field is eliminated. For advanced RP patients, objects are seen as if through a keyhole. Two images simulate the vision of an RP patient
To treat polygenic diseases such as AMD, gene therapy would need to identify a number of genes and be able to reintroduce them back to the patient’s genome effectively. Moreover, gene therapy is not effective in advanced stages of retinal degeneration after the target cells have degenerated. Therefore, developing the means to produce young cells that can replace aged cells has become an important goal for stem cell research in the retina.
Embryonic Stem Cells and Retinal Cells
Stem cells have the potential to differentiate into a number of cell types and are capable of extended self-renewal. There are several possible sources from which the cells can be derived. For instance, embryonic stem (ES) cells are stem cells derived from the inner cell mass of blastocyst-stage embryos. They consist of three germ cell layers, endoderm, mesoderm, and ectoderm, each with the potential to differentiate into a variety of cells.
In 2004, the Takahashi group described the differentiation of monkey ES cells into RPE by culturing ES cells with PA6 stromal cells in a differentiating medium [13]. The resultant ES cell-derived pigment epithelium expressed typical RPE markers and enhanced the survival of photoreceptors when grafted into the subretinal space of 4-week-old Royal College of Surgeons rats [13]. The generation of photoreceptors from mouse ES cells was more complicated, requiring culture under serum-free suspension conditions with Wnt and Nodal antagonists (Dkk1and LeftyA), then activin and serum (termed SFEB/DLFA-treated ES cells) [14]. These SFEB/DLFA-treated ES cells expressed Rx and Pax6, suggesting photoreceptor differentiation. They were able to integrate into explanted embryonic retinal tissue; approximately 10 % localized to the outer nuclear layer and possessed a protrusion on the outer side characteristic of photoreceptor outer segments [14].
More recently, several groups have described the generation of RPE and photoreceptors from human ES cells [15–19]. Vugler et al. [17] described the generation of RPE cells from human ES cells. The human ES-derived RPE cells were shown to express markers of developing RPE such as OTX1/2 and Pax6, as well as markers of mature RPE such as RPE65 [17]. When transplanted into the subretinal space of RCS rats, the cells maintained the expression of RPE65, while downregulating the expression of developing markers [17]. The same human ES-derived RPE cells were found capable of phagocytosing porcine outer segments in vitro and human photoreceptors in an artificial ex vivo human retina system [18]. Osakada et al. [19], also in 2008, generated RPE and photoreceptors from human ES cells by culturing human ES cells with Wnt and Notch inhibitors to produce RPE, and then adding retinoic acid and taurine to generate photoreceptors.
ES cells carry a risk of immune rejection because they do not have the same immune profile as the host. There are also ethical issues associated with the use of ES cells. Other potential sources of stem cells include fetal stem cells and adult stem cells. Fetal stem cells can be used for transplantation when they are allowed to differentiate into retinal cells but transplanted before they form any intrinsic connections. However, ethical issues are associated with fetal stem cells as well, and the supply of fetal stem cells is limited. Adult bone marrow and umbilical cord blood also contain stem cells capable of differentiating into retinal cells. The benefit of using cord blood or bone marrow stem cells is that they make immunosuppression after transplantation unnecessary (see Table 7.1). In addition, no gene correction is needed because these cells do not contain the culprit genetic mutations. An FDA approved safety trial at the Wills Eye Institute is currently underway to test for the use of cord blood or bone marrow stem cells in the treatment of macular disease.
Table 7.1
Diverse sources of stem cells: comparing research promise and challenges
Cord blood/bone marrow stem cells | Advanced cell technology’s embryonic stem cells trial at UCLA | Adult skin-derived stem cells (iPS) |
|---|---|---|
FDA approval | FDA approved safety trial showed promising results from one Stargardt and one macular degeneration patient. Vision was improved in both patients within a 4-month period Results from efficacy trial expected by 2015 | In development |
No immunosuppression, stem cell survival is transient | Immunosuppression similar to a kidney transplant regimen is required | No immunosuppression needed for adult skin derived stem cells since they are derived from patient |
No gene correction in stem cell necessary | No gene correction in stem cell necessary | Gene correction in stem cells may be necessary in many cases |
Induced Pluripotent Stem Cells and Retinal Cells
The use of ES cells in transplantation is limited by the possibility of immune rejection and other ethical issues. The development of induced pluripotent stem cells in 2006 has provided a way of producing pluripotent stem cells from somatic cells without the use of an oocyte, thereby avoiding immune rejection [20]. Takahashi and Yamanaka were able to induce pluripotency in somatic cells by the ectopic expression of four defined transcription factors: Oct4, Klf4, Sox2 and Myc [21, 22]. The most common source of somatic cells has been skin fibroblasts, but the risk of complications from using skin fibroblasts has led to the search for other sources of patient tissue [20].
More recently, induced pluripotent stem cells (iPSCs) have been derived from terminally differentiated T cells in the peripheral blood [23–25]. During this process, mononuclear blood cells were isolated and induced by integrating and non-integrating vectors such as lentiviruses, retroviruses, or temperature-sensitive mutated Sendai viruses into embryoid bodies containing endodermal, mesodermal, and ectodermal markers [23–25]. When injected subcutaneously into mice, the cells developed into tumors containing tissue from all three germ cell layers [23–25].
Clearly, the use of integrating vectors increases the potential for tumor formation and mutagenesis in the transplant’s host. Any cells treated with genes and vectors and then transplanted into human patients need to be assessed for tumorigenesis beforehand. In 2009, Hirami et al. [26] described the generation of human photoreceptors and RPE cells from iPSCs using Wnt and Notch inhibitors to produce RPE, and adding retinoic acid and taurine to generate photoreceptors. The use of iPSC-derived retinal cells brings us closer to true personalized medicine. Unfortunately, tumor formation is a risk of both ES cell and iPSC transplantation because not all stem cells differentiate using a differentiation protocol, and the non-differentiated cells are transplanted along with differentiated cells into the host retina, where the results are unpredictable.
Stem Cell Transplantation
Stem cell transplantation has the potential to provide treatments for the advanced stages of retinal degeneration with retinal neuron loss. However, for the transplant to succeed, the transplanted cells must integrate into the host retina through synapse formation. Photoreceptors should be able to integrate more readily than other types of neurons; they only need to create one synaptic connection with a second-order neuron, since their afferent responses depend not on receiving impulses from other neurons but from light. Meanwhile, RPE does not require reconnection with another neuron, although it does require integration with the Bruch membrane. Interestingly, grafted RPE do not require contact with the Bruch membrane in order to restore retinal function in animal models [27]. Moreover, a few transplanted cells can restore visual function globally.
Early transplantation experiments in rodents using whole sheets of fetal retina demonstrated that the transplanted cells only integrated into the host retina to a limited extent. The increase in visual function was thought to be largely due to the expression of trophic factors by grafted tissues that maintained survival of existing host photoreceptors. In 1999, human fetal neural retina tissue was transplanted into six patients with RP and four patients with AMD [28]. Visual acuity improved in 70 % of the patients. One patient maintained his improved visual acuity for 6 years. More recent studies of transplanted human fetal retinal tissue to patients with RP have not shown improvement in overall visual acuity [29]. Transplantation of fetal retinal tissue into patients with advanced AMD has even demonstrated graft rejection [30–32].
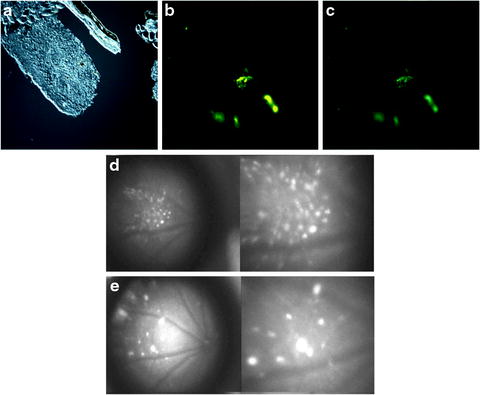
In contrast to fetal retina tissue transplantation, stem cell transplantation may have better integration potential in the host retina. Stem cells have greater plasticity and migrating capacity. A rat model of RP has shown rescue by a human RPE cell line (ARPE-19) and by RPE cells derived from human ES cells [33–36]. A human RPE cell-line (ARPE-19) improved retinal function in RCS rats [34, 35]. Similarly, human stem cell-derived RPE also improved visual function in RCS rats [17, 33, 36]. Most recently, iPSC-derived RPE improved retinal function in RCS rats [18]. In 2010, RPE derived from ES cells of C57BL/6J-Tyr c-2j /J (C2J) mice were transplanted into a spontaneous mouse mutant model of RP, termed the rd12 mice, on a C57Bl/6J background [37]. The RPE, labeled with yellow fluorescent protein, demonstrated survival of the graft within the host retina (see Fig. 7.4). Approximately one-fourth of the injected mice showed improvements in electroretinogram (ERG) responses after transplantation, demonstrating proper integration of graft tissue and functional rescue [37]. In 2009, photoreceptors derived from human ES cells were successfully transplanted into a mouse model of Leber congenital amaurosis [38]. In this study, transplanted cells integrated into the outer nuclear layer, expressed layer-specific opsin and rhodopsin, and restored the light reflex [38] (see Fig. 7.5).
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
