Fig. 5.1
Graves’ ophthalmopathy or thyroid eye disease (TED) (red circle) occurs in 90 % of patients in conjunction with Graves’ hyperthyroidism, but 10 % is euthyroid or hypothyroid. Patients with Graves’ hyperthyroidism (yellow circle) have clinically manifest TED in about 25–35 %, and subclinical TED in the majority of the remaining 65–75 %. Smoking is a main determinant of the transition from subclinical to overt TED
We will review the association between smoking and TED, the biologic mechanism behind this association and whether the association indicates a causal relationship. We also discuss if cessation of smoking could prevent TED, and explore smoking cessation strategies.
Association Between Smoking and TED
A meta-analysis published in 2002 concluded that smoking increases the risk of TED beyond the risk associated with Graves’ hyperthyroidism alone [10]. Comparison of smoking behaviour between patients with Graves’ disease (Graves’ hyperthyroidism, with cases of TED included in 5 of the 8 studies) and controls revealed current smoking as a risk factor for Graves’ disease with an odds ratio of 3.30; there was no increased risk among former smokers (Table 5.1). When comparing TED patients with controls, the risk for ever smokers was significantly higher with an odds ratio of 4.40. Restricting the analysis to patients with Graves’ hyperthyroidism with or without TED, current smoking significantly increases the risk of TED with an odds ratio of 2.18. Essentially similar results have been obtained in a systematic review published in 2008 [11]. The observed association between smoking and Graves’ disease is somewhat stronger in women than in men [10], and found in diverse ethnic groups [5, 10, 12–14]. In view of the undisputed association between smoking and TED, the question emerges whether smoking has a causal relationship with TED.
Table 5.1
Meta-analyses of studies on the risk of Graves’ disease associated with smoking
Odds ratio (95 % confidence interval) | |
|---|---|
Graves’ disease patients vs. controls | |
Current smokers vs. never smokers | 3.30 (2.09–5.22) |
Ever smokers vs. never smokers | 1.90 (1.42–2.55) |
Ex-smokers vs. never smokers | 1.41 (0.77–2.58) |
Graves’ ophthalmopathy vs. controls | |
Ever smokers vs. never smokers | 4.40 (2.88–6.73) |
Graves’ ophthalmopathy vs. Graves’ disease | |
Current smokers vs. never smokers | 2.18 (1.51–3.14) |
Ever smokers vs. never smokers | 2.53 (1.70–3.77) |
Several features of the association suggest causality [15]:
1.
Strength. There is some heterogeneity in the analysed studies, which differ in sample size, age and sex distribution, and definition of current smoking. Also, some studies do not discriminate between Graves’ hyperthyroidism and TED, labelling all patients as Graves’ disease. Most studies are cross-sectional with just a few being prospective. Despite these deficiencies, strong associations (high odds ratio) are found.
2.
Consistency. The likelihood of a causal relationship increases if the association is observed in different populations. This certainly is true for TED: studies from various countries in Europe, Asia and the USA all identified smoking as a risk factor [10].
3.
Dose–response relationship. A Japanese study describes an increased risk of Graves’ hyperthyroidism with an increasing number of cigarettes smoked per day (odds ratio 3.7 for 1–10 cig./day and 5.1 for 21–40 cig./day, p < 0.01 for trend) [13]. In a UK study among patients with Graves’ hyperthyroidism, the proportion of patients with TED increased as cigarette consumption increased (p = 0.008) [12]. Many studies describe a dose–response relationship between current smoking and the severity of TED [10, 12]. In a retrospective survey the frequency of current smokers with moderate, severe and malignant TED was 64 %, 71 % and 87 %, respectively [16]. In another study smokers had more severe eye disease than nonsmokers as assessed by the total eye score (12.2 ± 6.9 vs. 8.6 ± 6.2; p = 0.03), and patients with more severe forms of TED were more often smokers than those with no or less severe TED resulting in a significant increase in the odds ratio (p < 0.001 by trend test) (Fig. 5.2) [17]. In a prospective study there was a significant increase in the relative risk for proptosis with increased smoking; the relative risk for diplopia was 1.8 for patients currently smoking 1–10 cigarettes daily, 3.8 for those smoking 11–20 cigarettes and 7.0 for patients smoking >20 cig./day [18]. In this study lifetime tobacco use was not an independent risk factor for the development of TED symptoms. A study from Korea reports that smoking status was a predictive risk factor for a severe course of TED and the development of optic neuropathy (odds ratios 6.57 and 10.0, respectively) [14]. The data support a quantitative relationship between current smoking and the severity of TED: the higher the number of daily cigarettes, the greater the severity of TED.
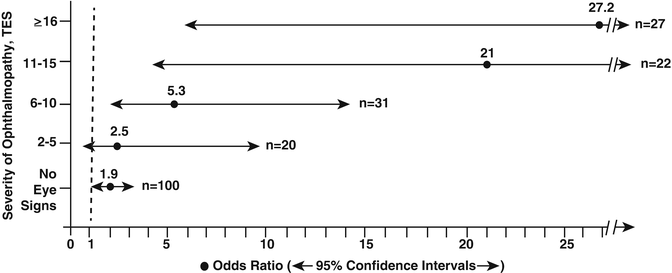
Fig. 5.2
The increase in the prevalence of smokers (represented by the odds ratio with 95 % confidence intervals) in relation to the severity of TED (assessed by the total eye score TES) in patients with Graves’ hyperthyroidism. From Prummel MF, Wiersinga WM. Smoking and risk of Graves’ disease. JAMA 1993; 269: 479–482. Reprinted with permission from American Medical Association
4.
Temporality. To stop smoking apparently decreases the risk for Graves’ disease, as the odds ratio is lower in ever smokers than in current smokers (but still significant) while it is no longer significant in former smokers (Table 5.1). In the prospective study by Pfeilschifter et al. [18], the incidence of TED among current smokers, former smokers and never smokers was 68.2 %, 64.0 % and 51.7 %, respectively; the incidence of proptosis in these groups was 49.0 %, 16.0 % and 18.6 %, and that of diplopia 27.9 %, 16.0 % and 8.9 %, respectively. The risk for TED was not increased in former smokers compared to never smokers (TED incidence RR 1.45, 95 % CI 0.90–2.33; proptosis incidence RR 0.90, 95 % CI 0.24–2.33; diplopia incidence RR 1.88, 95 % CI 0.46–7.73). When current smokers were subdivided in those smoking 1–10 cig./day, 11–20 cig./day and >20 cig./day, the incidence of proptosis in these three groups was 36.7 %, 59.5 % and 62.5 %, respectively; the incidence of diplopia in these groups was 16.3 %, 34.0 % and 62.5 %, respectively. Cessation of smoking may lead to a lower incidence of TED and to less severe TED.
5.
Specificity. Is the risk of smoking specific for hyperthyroidism when it is caused by Graves’ disease? Apparently so, because ever smoking was not associated with hyperthyroidism due to toxic nodular goitre (odds ratio 1.22, 95 % CI 0.96–1.55) [10]. Is the risk of smoking specific for Graves’ disease and not for other autoimmune thyroid diseases? It appears so because recent epidemiological studies have demonstrated that smoking is associated with a lower occurrence of TPO- and Tg-antibodies and the development of overt autoimmune hypothyroidism (Hashimoto’s disease) [19, 20]; cessation of smoking transiently increases the risk of developing overt hypothyroidism [21]. The contrasting effects of smoking on the risk of Graves’ and Hashimoto’s disease resemble the divergent effects of smoking on inflammatory bowel disease: smoking is a risk factor for Crohn’s disease (odds ratio 1.61, 95 % CI 1.27–2.03), but has a protective effect for colitis ulcerosa (odds ratio 0.41, 95 % CI 0.34–0.48) [22]. Smoking is also a known risk factor for other autoimmune diseases like rheumatoid arthritis and systemic lupus erythematosus. Smoking is thus a disease modifier of many autoimmune conditions, but its consequences appear to be rather disease specific.
6.
Biological plausibility. The studies reporting the association between smoking and Graves’ disease might have been affected by confounding factors like iodine intake, stress exposure and alcohol intake. While possible, this seems unlikely for several reasons. First, the association has been observed in areas with iodine deficiency as well as in areas with sufficient iodine intake. Second, smoking behaviour might be related to stress, but smoking was still an independent risk factor for Graves’ disease after adjustment for stressful life events [13]. Third, smoking behaviour might be connected with alcohol intake, but alcohol consumption has a protective effect on both Graves’ hyperthyroidism and Hashimoto’s hypothyroidism and its effects are independent of smoking [23, 24]. Confounding factors can never be excluded completely, but the association between smoking and TED apparently is with exposure to cigarette smoke itself and not with a factor connected with smoking behaviour. Although the pathogenesis of TED is still incompletely understood, a number of in vivo and in vitro studies report how smoking might favour the development of TED. This is discussed in the next section.
Taken together, the strong and consistent association between smoking and TED, the presence of a dose–response relationship, the temporality of the risk (which decreases after cessation of smoking), the specificity of the risk for Graves’ disease and not for Hashimoto’s disease and the biological plausibility of the association, all support the existence of a causal relationship between smoking and TED.
Biological Mechanisms Relating Smoking to TED
1.
Immunopathogenesis of TED: A synopsis [25, 26]. The hallmark of TED is swelling of extraocular muscles and orbital fat. It is caused mostly by excessive secretion of glycosaminoglycans (very hydrophilic compounds which attract a lot of water) by activated orbital fibroblasts, and differentiation of a subset of orbital fibroblasts into mature adipocytes. Orbital fibroblasts have been recognized as the target cells of the orbital autoimmune attack in Graves’ disease; they express low levels of TSH receptors, which however markedly increase upon differentiation into mature fat cells. Homing of immunocompetent cells to the orbit is facilitated by cytokine-induced expression of HLA and adhesion molecules on vascular endothelium. Graves’ immunoglobulins induce orbital fibroblasts to secrete T cell chemoattractants. The numerous macrophages but also the few B cells in the orbit may act as antigen-presenting cells that in the setting of MHC class II molecules present linear TSHR peptides to T cells. Activated T cells release a variety of inflammatory cytokines, chemokines and prostaglandins, causing inflammatory edema and proliferation of fibroblasts. The cytokine profile in the early stages of TED is predominantly derived from Th1-cells, inducing orbital fibroblasts to secrete cytokines and excessive amounts of glycosaminoglycans, which cause swelling of orbital tissues, in particular of extraocular muscles. A subset of fibroblasts differentiates into adipocytes under the influence of particular cytokines. In late stages of TED, the cytokine profile is mostly Th2-cell derived and the effect of Graves’ IgG becomes more prominent in the process of adipogenesis. TSHR antibodies bind to TSHR on orbital fibroblasts, resulting in upregulation of hyaluronan synthase and excessive production of hyaluronan, one of the prevalent glycosaminoglycans in the orbit.
2.
Effect of smoking on immune responses in general. The immunological effects of cigarette smoking have been studied extensively. However, it remains difficult to relate changes in the number of immunocompetent cells and the concentration of their products in the blood to functional alterations in other immunological compartments, and then to link these alterations to specific diseases. Adding to the complexity are the many components of cigarette smoke; some of these over 4,800 compounds are endotoxin (acting via Toll-like receptor 4), nicotine (acting via nicotinic acetylcholine receptor), reactive oxygen species/free radicals (acting via NOD-like receptor family, pyrin domain containing 3) and polycyclic aromatic hydrocarbons (acting via aryl hydrocarbon receptors) [27]. It has become clear that smoking affects both the innate and adaptive immune systems [28]. Cigarette smoke activates innate immune cells (like macrophages, dendritic cells, neutrophils and epithelial cells) via oxidative stress and by triggering pattern recognition receptors (PPR, such as Toll-like receptors, NOD-like receptors and purinergic receptors). The activation of PPR may occur directly by cigarette components or indirectly via the release of damage-associated molecular patterns (DAMP) by dying—autophagic death, apoptotic or necrotic—cells [27]. DAMPs can be released from injured cells due to exposure to tobacco smoke, oxidative stress, infections or tissue hypoxia. DAMPs released from stressed or dying cells include uric acid, heat shock proteins, β-defensins and interleukin-1α, among many others, and DAMPs released from the breakdown of extracellular matrix encompass glycosaminoglycans like hyaluronan, heparin sulphate, versican and biglycan (acting via Toll-like receptors) and fibronectin (acting via Toll-like receptor-4 and integrins). Following stimulation by PPR, the innate immune cells release pro-inflammatory cytokines and chemokines, reactive oxygen species (ROS) and proteolytic enzymes, such as neutrophil elastase and matrix metalloproteinases. Activated dendritic cells induce adaptive immune responses involving T helper (Th1 and Th17) CD4+ T cells, CD8+ cytotoxicity and B-cell responses. Cigarette smoke increases the production of many pro-inflammatory cytokines, such as TNF-α, Il-1, IL-6, IL-8, and decreases the production of anti-inflammatory cytokines such as IL-10 [28]. Smoking also induces oxidative stress, enhances the generation of free radicals, and decreases the level of antioxidants not only in the blood but also in aqueous humour and ocular tissue [29, 30].
3.
Effect of smoking on immune responses in TED: in vitro studies. Cultures of human orbital fibroblasts have been used to explore how smoking might affect the immune response in TED. In the absence of a validated animal model of TED, the rationale of using orbital fibroblasts as a surrogate experimental model is that orbital fibroblasts on the one hand are the target cells of the orbital autoimmune attack and on the other hand also effector cells by their production and secretion of excessive amounts of glycosaminoglycans. Smoking reduces tissue oxygen tension by causing vasoconstriction and the formation of carboxyhemoglobin. Extraocular muscle fibroblasts when cultured under hypoxic conditions (5 % CO2: 95 % N2 vs. 5 % CO2: 95 % air in normal conditions, resulting in PO2 of 13 and 18.6 kPa, respectively), display a significant increase in glycosaminoglycan, protein and DNA synthesis, under both basal and cytokine-treated conditions [31]. Incubation of orbital fibroblasts with H2O2 increases the expression of heat shock protein-72 (HSP-72) [32]. Cultured orbital fibroblasts obtained from patients undergoing orbital decompression for severe TED did not express HLA-DR when treated with nicotine or tar; HLA-DR expression increased about threefold upon exposure to nicotine or tar in combination with interferon-γ [33]. Cigarette smoke contains large quantities of superoxide radicals and other ROS. Exposure of orbital fibroblasts to superoxide radicals, induced by adding xanthine oxidase and hypoxanthine to the culture medium, caused a dose-dependent proliferation of fibroblasts derived from TED patients but not from control fibroblasts; proliferation was inhibited by the xanthine-oxidase inhibitor allopurinol, the antioxidant nicotinamide and by methimazole [34]. In another experiment cultured orbital fibroblasts were exposed to cigarette smoke extract (CSE) [35]. Fibroblasts from TED patients and controls showed similar results. CSE did not affect ICAM1 (intercellular adhesion molecule-1) expression. CSE did stimulate hyaluronic acid production in a dose-dependent manner, with 5 % CSE causing an increase of 44 % (p = 0.001); 5 % CSE compares to 40 cigarettes daily. CSE also increased adipogenesis in a dose-dependent manner, as did interleukin-1; the effect of 5 % CSE is approximately half that seen with 0.1 ng/mL IL-1. The effects of CSE and IL-1 on adipogenesis were synergistic: the degree of adipogenesis in the well containing 5 % CSE and 0.1 ng/mL IL-1 was double the magnitude of the sum of the values obtained from either stimulus alone, and reduced by 82 % by addition of an anti-IL-1 antibody. Lastly, CSE and H2O2 (used as a control) dose-dependently stimulated intracellular ROS production and adipogenesis in orbital fibroblasts from TED patients and control subjects [36]. The effects of 2 % CSE and 10 μM H2O2 were similar. Addition of quercetin, a flavonoid phytoestrogen with antifibrotic properties, inhibited adipogenesis by reducing ROS generation in vitro. Quercetin thus holds promise as a therapeutic option in TED patients [37, 38].
Taken together, the results with cultured orbital fibroblasts strongly support the notion that exposure to cigarette smoke is causally connected with TED. Oxidative stress plays a major role in TED as several studies have indicated greater oxidative DNA damage, lipid peroxidation and ROS production in cultured orbital fibroblasts from TED patients relative to normal orbital fibroblasts [39–41]. Cigarette smoking greatly increases oxidative stress. It induces expression of HLA-DR and HSP-72 on orbital fibroblasts, which is involved in antigen recognition and T cell recruitment. It causes orbital fibroblasts to proliferate, to produce excessive amounts of glycosaminoglycans and to differentiate into adipocytes. Many of these effects of smoking are dose dependent, and may occur or are potentiated in a particular cytokine environment. Smoking-induced hypoxia may further aggravate these events.
4.
Effect of smoking on immune responses in TED: in vivo studies. Several studies have investigated whether smokers differ from nonsmokers with respect to serum concentrations of adhesion molecules, cytokines and TSHR antibodies. TED patients had higher levels of the adhesion molecules sICAM-1, sVCAM-1 and sELAM-1 than Graves’ disease patients without TED or healthy controls; only sICAM-1 correlated with the severity of TED. Smoking was independently associated with higher levels of sICAM-1 and lower levels of sVCAM-1 in both TED patients and controls; it did not affect sELAM-1 [42]. Serum concentrations of IL-1α, IL-1β and TNFα were not influenced by smoking or the presence of TED [43–45]. Soluble interleukin-1 receptor antagonist (sIL-1RA) levels, however, were lower in smokers than in nonsmokers with TED; baseline sIL-1RA and its increase during immunosuppressive treatment were lower in nonresponders than in responders to therapy [43]. It was suggested that measurement of sIL-1RA might predict the therapeutic response. However, subsequent studies could not confirm these findings since they reported similar sIL-1RA concentrations between smokers and nonsmokers and between responders and nonresponders to immunosuppression in TED patients [44–47]. Serum sIL-2R, IL-6, IL-6R, TNFαRI, TNFαRII and sCD30 concentrations were all higher in TED patients than in healthy controls, but levels did not differ between smokers and nonsmokers [47]. Serum IL-6 and IL-6R are higher in active than in inactive TED [44].
Serum concentrations of IL-1β, s IL-1RA, TNFα, IL-6 and sIL-6R were not different in Graves’ disease patients with or without TSHR antibodies [44]. Although the serum concentration of TSHR antibodies is higher in TED than in Graves’ patients without TED, the levels are not different between smokers and nonsmokers [16, 48]. No correlation was found between smoking (as assessed by urinary levels of cotinine, the major metabolite of nicotine) and the level of TSHR antibodies in TED [49]. Another study in TED patients likewise found no difference in TSHR antibodies between smokers and nonsmokers [50]. Serum concentrations of TSHR antibodies are higher in patients with a severe course of TED compared with patients with a mild course of TED, but this relationship of TSHR antibodies to TED severity was independent from smoking [51].
An early report distinguished TED patients without restrictive myopathy (type I) and with restrictive myopathy (type II); 63 % of type I and 83 % of type II were smokers [52]. The data suggest smoking affects enlargement of extraocular muscles to a greater extent than enlargement of the orbital fat/connective tissue compartment. A subsequent volumetric study, however, observed a direct relationship between orbital connective tissue volume and current smoking, whereas extraocular muscle volume was not influenced by smoking [53]. In contrast, a recent study in consecutive-untreated TED patients found that smoking did not influence orbital fat volume, but that smokers had significantly larger extraocular muscle volumes than nonsmokers [54].
Taken together, the higher serum concentrations of some adhesion molecules and cytokines in smokers as well as the differential effect of smoking on orbital fat and muscle enlargement provides some evidence regarding the mechanism by which smoking is causally related to TED.
Smoking and Prevention of TED
The concept of prevention is illustrated in Fig. 5.3. Primary prevention aims at keeping the disease from occurring at all by removing risk factors. Secondary prevention is directed at early detection when the disease is still asymptomatic and when early treatment can stop the disease from progressing. Tertiary prevention refers to those clinical activities that prevent further deterioration or reduce complications after the disease has declared itself [55].
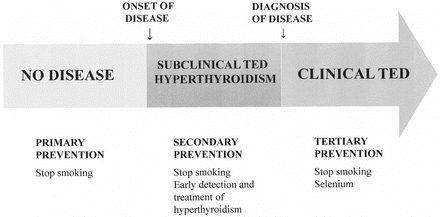
Fig. 5.3
Primary, secondary and tertiary prevention of TED
1.
Get Clinical Tree app for offline access

Primary prevention. The biological plausibility of a causal relationship between smoking and TED suggests that to refrain from smoking may diminish the chance to develop both Graves’ disease and TED. This notion is strongly supported by the significant odds for current smokers to contract Graves’ disease, whereas the odds are lower in ever smokers and not significant in former smokers [10]. Indeed current, but not lifetime, tobacco consumption constitutes a risk for the incidence of proptosis and diplopia in Graves’ patients [18].
Indirect evidence for the efficacy of smoking cessation as primary prevention of TED comes from questionnaire studies. A European survey in 1996 revealed that 43 % of physician respondents thought TED was decreasing in frequency, and 12 % thought it to be increasing [56]. The supposed decrease most likely was attributed to a secular trend towards a lower prevalence of smoking in most European countries. It is remarkable that all respondents from Hungary and Poland (where the proportion of smokers in the general population had increased since the fall of the Berlin Wall) indicated an increased incidence of TED. Another European questionnaire survey collected data on pediatric Graves’ disease [57]. The prevalence of TED among children with Graves’ hyperthyroidism was 30 %. When grouped according to smoking prevalence among teenagers in the country of origin, it became evident that TED frequency was the highest in countries in which teenagers smoked most (Table 5.2). Divided by age, the highest proportion of children with TED 10 years of age or younger was found in countries where the smoking prevalence was highest. As it is unlikely that children ≤10 year smoke, the results are best explained by passive smoking as a result of living in an environment in which 25 % or more of their peers smoke.
Table 5.2




Frequency of childhood TED in pediatric Graves’ hyperthyroidism as a function of smoking prevalence among teenagers in their country of origin
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree