This chapter is dedicated to Maurice Rabb, M.D. (1932–2005), previous author of this chapter. Dr. Rabb devoted his life to the research and care of patients with sickle cell disease.
In Western literature, the first description of the clinical manifestations of sickle cell anemia was published by Herrick1 in 1910; he noted the sickled appearance of red blood cells (RBCs) in a young student from the West Indies. It was not until 1930, however, that the first report of retinal changes in sickle cell disease appeared. Cook2 described a retinal hemorrhage in a young patient. A great deal is now known about the multiple ocular manifestations of sickle cell disease, which may be seen in the orbits, anterior segment (conjunctiva, anterior chamber, iris), retina, choroid, and optic nerve. These changes result from vascular occlusions caused by sickled RBCs and from the increased adhesion of these cells to the vascular endothelium. The visible vascular networks of the eye provide the clinician a unique opportunity to directly observe the vaso-occlusive process. A brief review of the genetics and pathophysiology of sickle cell disease is followed by a detailed description of the clinical manifestations and management of sickle cell eye disease.
GENETICS AND EPIDEMIOLOGY
The hemoglobin (Hb) molecule is composed of two pairs of polypeptide chains: α and β. Two α-genes and one β-gene, located on chromosome 16 and chromosome 11, respectively, encode these chains. An individual may have two different types of β-chains, while there are four α-chain variants. However, some individuals, most commonly in people of African origin, have a chromosome 16 that contains only one α-gene (one is deleted). Hemoglobin A (Hb A) and hemoglobin A2 (Hb A2) are the predominant types of hemoglobin found in postnatal life. Hb A contains two α-chains and two β-chains; Hb A2 contains two α-chains and two δ-chains. Fetal hemoglobin (Hb F), predominant during prenatal life, contains two α-chains and two γ-chains. A developmental gene switch from expression of fetal γ-globin to adult β-globin occurs on the basis of gestational age. After the end of the first year, RBC composition typically remains stable.3
In 1917, Emmel4 first suggested a genetic basis for the sickling of RBCs, and by 1923, it was clear that this was inherited in a Mendelian pattern.5 By 1933, symptomatic cases (termed sickle cell anemia) were differentiated from asymptomatic, latent cases (termed sickle cell trait).6 In 1949, using electrophoresis, Pauling et al.7 discovered that sickle cell disease was caused by the presence of an abnormal hemoglobin molecule, named sickle hemoglobin (Hb S). This abnormal hemoglobin imparts a sickle shape to deoxygenated RBCs. In 1956, it was reported that Hb S differed from Hb A by a single amino acid substitution (i.e., valine for glutamic acid) in the sixth position from the N-terminal end of the β-chain.8, 9 This amino acid substitution results from a single DNA base point mutation in which thymidine is substituted for adenine in the sixth position of the β-gene (i.e., GAG to GTG). Several other point mutations have been described; another important variant is hemoglobin C (Hb C), caused by the substitution of lysine for glutamic acid in the sixth position of the β-chain.
Thalassemias are genetic variations in hemoglobin genes that result in abnormal or absent globin chain synthesis. They are classified according to the type and amount of globin chain present. Synthesis of either of the two types of polypeptide chains in the hemoglobin molecule may be affected, resulting in either α-or β-thalassemia. β-Thalassemia may have no β-chain present (β0-thalassemia) or a reduced amount of β-chain present (β+-thalassemia); in α−-thalassemia, one or more α-globin genes are absent or nonfunctional, leading to reduction of α-chain synthesis.10
The term hemoglobinopathy describes genetic disorders associated with structurally abnormal hemoglobin or subnormal production of hemoglobin. Sickle cell disease is the most common hemoglobinopathy affecting humans, with a gene frequency as high as 45% in equatorial Africa.11 Hemoglobinopathies occur in a heterozygous or homozygous form. Approximately 8% of African-Americans are heterozygous for the gene for Hb S, known as sickle cell trait (Hb AS), in which the RBC contains normal Hb A together with abnormal Hb S, usually in a 55:45 ratio.10 Sickle cell trait is usually asymptomatic and is not thought to affect life expectancy. The homozygous state, Hb SS, results in sickle cell disease, termed Hb SS disease. However, other heterozygous states, such as compound heterozygosity for Hb S/Hb C, can also cause clinical sickle cell disease, termed Hb SC disease.
The systemic manifestations of sickle cell disease include anemia, organ infarcts in the lungs, spleen, kidney, and liver, and pain crises in the limbs and trunk. While the genetic mutation is single, the clinical presentation is highly variable. The expression of the disease depends on the amount of Hb F present in postnatal life and the presence or absence of α-thalassemia. The presence of other point mutations in the hemoglobin gene or in other gene regions closely associated with the locus for the hemoglobin β-chain (haplotypes) may be capable of modulating the expression of the gene as well.10 Sickle cell trait (Hb AS) is generally excluded from the definition of sickle cell disease because of its minimal clinical manifestations. However, infrequently patients with sickle cell trait may develop ocular complications.
The hemoglobinopathies that produce sickle cell disease are Hb SS, Hb SC, and Hb S-β−-thalassemia (β0 or β+). Hb SS disease, or homozygous sickle cell disease, is associated with the most severe systemic manifestations and often early morbidity. Hb SC disease, or doubly heterozygous sickle cell disease, is associated with milder systemic manifestations than Hb SS disease but is associated with more serious retinal disease. In the United States, the prevalence of Hb SC disease is 1 in 1,500, compared with 1 in 625 of Hb SS disease.11 The most significant sickle cell-thalassemia disease is Hb S-β−-thalassemia, the clinical symptoms of which depend on how much Hb A β-chain is present and whether there is high expression of Hb F, which has an ameliorating effect on sickle cell disease. Three other less common genotypes manifesting the features of sickle cell disease include Hb SD Punjab, Hb SO Arab, and Hb S Lepore Boston. Hb D has a glutamine residue instead of a glutamic acid residue at position 121 on the β-chain. Hb O has a lysine instead of glutamic acid, also at position 121 on the β-chain.12
Restriction endonuclease analysis suggests that the Hb S gene arose from three geographically independent mutations in equatorial Africa.13, 14 From Africa, the gene was carried to North and South America, the Carribean, and Europe. The sickle cell gene independently appeared around the Mediterranean (Italy, Greece), Middle East (Saudi Arabia), and India. Surprisingly, the sickle gene frequency has remained relatively stable.14, 15 One theory put forth to explain the evolutionary survival of a gene mutation with such devastating clinical manifestations and early morbidity is called balanced polymorphism. According to this theory, the negative effects of a genetic mutation are balanced by its protective benefits, which aid in natural selection. In this case, the Hb S gene, when it occurs in the heterozygous state, offers some benefit by protection against malaria.16, 17 The greatest prevalence of the Hb S gene exists in the malarial regions of Africa, and children with sickle cell trait (Hb AS) are afforded some degree of protection against malaria, particularly the type caused by Plasmodium falciparum.18
Like the Hb S gene, the Hb C gene is also highly concentrated in West Africa and offers some protection against malaria.14 Approximately 2% of African-Americans carry one gene for Hb C, mostly as C trait (Hb AC). Homozygous C (Hb CC) is rare, occurring in 0.016% of African-Americans. Homozygosity for Hb C does not affect life expectancy but is sometimes associated with mild anemia, a slightly enlarged spleen, and gallstones.
PATHOPHYSIOLOGY
Animal models have provided a means for in vivo studies of sickle cell disease. The first transgenic mouse bearing a human Hb S gene was developed in 1990 and simulated sickle cell trait.19, 20 The knockout transgenic mouse was developed in 199721, 22 and synthesized 100% Hb S, simulating sickle cell disease. These models have contributed significantly to the understanding of the pathophysiology of sickle cell disease.
RBCs containing Hb S acquire a sickle-shaped deformity upon deoxygenation, due to the linear aggregation of hemoglobin and subsequent polymer formation. Hb C forms polymers only in the presence of Hb S, whereas Hb F inhibits polymer formation. Damaged RBC membranes lead to potassium loss and intracellular dehydration, which further potentiates Hb S polymerization. Eventually, the RBC membrane is no longer capable of assuming the normal biconcave shape upon reoxygenation, thus forming an irreversibly sickled cell (ISC).
Oxygen is the most important determinant of Hb S polymerization. Very small decreases in arterial oxygen tension, even with an oxygen saturation greater than 90%, can result in sickling.23 Other factors that influence polymer formation are rate of deoxygenation, RBC age, intracellular Hb S concentration, decreased pH, and temperature. In the terminal arteriole and capillary circulation, oxygen availability decreases and the pH drops, enhancing Hb S polymerization.
Rheologic impairment underlies the complications of sickle cell disease.24 Vascular beds with low flow and high oxygen extraction are more prone to sickling and secondary vascular occlusion. The peripheral retina and macula appear to be the most susceptible to vascular occlusion.25 Interestingly, the terminal capillary bed in each of these zones borders on an avascular area and thins to a two-dimensional capillary bed.26 Small changes in blood vessel diameter dramatically affect the blood flow because flow resistance is inversely proportional to the fourth power of the vessel radius.27 The vascular occlusions of sickle cell retinopathy occur in the arterioles rather than in the capillaries, perhaps because the sphincters of the precapillary arterioles are narrower than the true capillaries.28, 29
Compared with normal cells, RBCs containing Hb S demonstrate greater vascular endothelium adherence. The nature of this adhesion is now known to be enormously complex, involving multiple cofactors, cytokines, receptors, and adhesive binding proteins. For example, CD36 is expressed on microvascular endothelium, and its receptor is on the RBC. The CD36 receptor is more exposed in RBCs containing Hb S. Other receptors that may be involved in adhesion include α4β1-integrin, membrane band 3, and sulfated glycoproteins. This adherence property is not exhibited by ISCs because their rigidity renders them unable to form a large surface contact with the endothelial cells. Thus, patients who generate a large number of ISCs have vascular occlusions at the precapillary arterioles because the rigid blood cells cannot enter the capillaries. Patients with fewer ISCs and more deformable cells have blood flow compromise in the capillaries, where RBCs containing Hb S readily adhere to the vascular endothelium.
There is an inverse relationship between the severity of systemic disease and the severity of sickle cell retinopathy in homozygous (SS) versus doubly heterozygous (SC) sickle cell disease. Patients with Hb SS disease have more systemic complications, with multiple vaso-occlusive events and secondary organ damage. Patients with Hb SC disease have fewer systemic complications but a greater frequency and earlier onset of retinal neovascularization, resulting in more severe sickle cell retinopathy and more visually disabling ocular complications. The prevalence of proliferative sickle cell retinopathy (PSR) increases with age in Hb SS and Hb SC disease (Tables 17-1 and 17-2).
The inverse relationship between the severity of systemic disease and the severity of sickle cell retinopathy in Hb SS versus Hb SC sickle cell disease may be explained by a discussion of the hematologic factors involved. In Hb SS disease, accelerated destruction of RBCs results in a state of marked anemia despite the common presence of brisk reticulocytosis.30, 31 Since the viscosity of blood is proportional to the hematocrit, the reduced hematocrit associated with Hb SS may protect against vascular occlusion. In contrast, patients with Hb SC and Hb S-β-thalassemia have higher hematocrits, causing higher whole blood viscosity and a relatively increased frequency of vascular occlusions in the retina. In addition, red cells from patients with Hb SC also have a higher mean corpuscular hemoglobin concentration, which also reduces cell deformability.
TABLE 17-1 Prevalence of Proliferative Sickle Retinopathy
Genotype
% with PSR
Hb SC disease
36
Hb S-β+-thalassemia
18
Hb S-β0-thalassemia
9
Hb SS disease
12
Modified from Condon PI, Hayes RJ, Serjeant FR. Retinal and choroidal neovascularization in sickle cell disease. Trans Ophthalmol Soc U K. 1980;100:434.
TABLE 17-2 Influence of Age on the Prevalence of Proliferative Sickle Retinopathy
Genotype
% with PSR, 8-17 y old
% with PSR, over 40 y old
Hb SC disease
11.0
68.0
Hb S-β+-thalassemia
NA
NA
Hb SS disease
0.4
14.0
NA, not available.
Modified from Penman AD, Talbot JF, Chuang EL, et al. New classification of peripheral retinal vascular changes in sickle cell disease. Br J Ophthalmol. 1994;78:681.
Analysis of hematologic factors, however, has so far provided only a partial explanation for the genotypic differences in the development of PSR. In a group of patients with homozygous sickle cell anemia, a significant relationship was found between the development of retinal neovascularization and high Hb levels and low Hb F levels in men, but not in women.32 In patients with SC disease, a significant relationship was demonstrated between the development of neovascularization and high mean cell volume in men and a low Hb F level in both men and women.33
Genotypic differences in the incidence of retinal neovascularization cannot be explained by the frequency of vascular occlusions alone: the more severe systemic complications of patients with homozygous sickle cell anemia are, in fact, a result of an increased frequency of vascular occlusions. α-Globin gene number does appear to reduce the extent of peripheral retinal vessel closure but has no apparent influence on the development of neovascularization in patients with homozygous sickle cell anemia.34, 35 In a study of the Jamaican Sickle Cohort, consisting of 173 Jamaican children with Hb SC disease, 315 with Hb SS disease, and 250 age- and gender-matched normal (Hb AA) controls recruited between 1973 and 1981, Talbot et al.36, 37 and 38 found that the peripheral retina demonstrates earlier vascular occlusions in Hb SS disease than in Hb SC disease. Comparing the peripheral retinal vascular bed with that of a normal cohort, however, they discovered that a significantly larger proportion of Hb SC disease subjects had an abnormal peripheral vascular pattern, which they were able to correlate with the subsequent development of neovascularization. The authors concluded that a normal border, even if undergoing a posterior regression, results from a gradual modification of the capillary bed and indicates a low risk for PSR, whereas an abnormal border occurs as a radical alteration of retinal perfusion.39 This cohort study also revealed that in Hb SS disease, vascular closures occurred more frequently with low Hb F levels, low mean total hemoglobin levels, high reticulocyte counts, and high ISC counts. In patients with Hb SC disease, closure was associated with high reticulocyte counts and lower height and weight.36, 37
The theory of ischemia versus infarction provides another explanation for the more severe PSR associated with Hb SC disease. The retinal vascular occlusions caused by RBCs in Hb SS disease might result in a more complete vascular occlusion and retinal infarction than in other hemoglobinopathies, causing necrosis of the retinal tissue. The less severe or more easily reversible vascular occlusions in Hb SC disease might result in retinal ischemia, which may provide an environment in which an angiogenic response, presumably by upregulation of vascular endothelial growth factor, is continuously stimulated. This theory is supported by the development of neovascularization in other disease processes associated with retinal ischemia, such as diabetes mellitus and retinal vein occlusion. Additional support of this theory is provided by the regression of neovascular tissue achieved by photocoagulating the ischemic retina around a neovascular membrane.40
Peachey et al.41, 42 studied the electroretinographic response in patients with sickle cell disease and neovascularization and correlated this with the degree of peripheral retinal capillary nonperfusion as determined by fluorescein angiography. They found significant correlations between reductions in electroretinographic amplitudes and the extent of retinal capillary nonperfusion in patients both with and without neovascularization. Patients with neovascularization also had prolonged generation of a maximum-amplitude response, similar to that seen in central retinal vein occlusion and diabetic retinopathy, suggesting that neovascularization is associated with the presence of ischemia in sickle cell retinopathy.
LABORATORY EVALUATION
The laboratory testing of patients with sickle cell disease is divided broadly into testing for the presence of Hb S, identifying the major genotypes, and subdividing the genetic characteristics of the major genotypes. For clinical purposes, it is not necessary for the ophthalmologist to subdivide the major genotypes; however, it is helpful to identify the major genotypes because they have different systemic and ocular clinical characteristics and prognosis.
The solubility test (Sickledex: Orthodiagnostics, Raritan, NJ) and the sickle prep test (metabisulfite slide test) are used to identify the presence of Hb S. The solubility test is based on the insolubility of deoxygenated Hb S, and the sickle prep test is based on the morphologic change of RBCs containing deoxygenated Hb S. The solubility test is simpler to perform and has gained widespread acceptance as a screening test. The major limitation of both tests is their inability to differentiate sickle cell trait from the clinically significant homozygous (SS) and heterozygous (SC) sickle cell disease. Therefore, a positive sickle prep test or solubility test must be followed by quantitative hemoglobin electrophoresis. The electrophoretic pattern of Hb SS and Hb S-β0-thalassemia are similar; however, they can be differentiated by quantification of Hb A2, which is elevated in Hb S-β0-thalassemia, but not in Hb SS disease. Sickle cell trait (Hb AS) can be distinguished from Hb S-β+-thalassemia since Hb S comprises more than 50% of the hemoglobin in Hb S-β+-thalassemia but less than 50% in sickle cell trait. DNA analysis by restriction endonuclease is the most widely used method for antenatal diagnosis of sickle cell disease, which may be accurately diagnosed as early as the second month of gestation.
CLINICAL OCULAR MANIFESTATIONS
Characteristic ocular findings result from the abnormal globin chain that, under certain conditions such as hypoxia, causes sickling of red blood cells and obstruction of blood vessels. Although the retinal manifestations of sickle cell disease are the most sight-threatening, pathology in the orbit, anterior segment (conjunctiva, anterior chamber, iris), and optic nerve occur as well.
Orbital
While bone infarction involving long bones is common in sickle cell disease, orbital involvement is rare. Thirty-two cases of orbital infarction, with or without subperiosteal hematomas, are described in the literature to date (Table 17-3).43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60 and 61 Dixit et al.62 reported a case in which the orbital compression syndrome was the presenting manifestation of a previously undiagnosed Hb S-β-thalassemia patient.
The pathophysiology of the orbital compression syndrome has recently been recognized. Infarction of the orbital bones (maxilla, zygoma, frontal) causes the initial insult, with a resultant inflammatory reaction. Local vessel wall necrosis and subsequent extravasation of blood can cause subperiosteal hematomas. The orbital inflammation and/or hematoma leads to compression within the orbit. The patient typically presents with rapidly progressive periorbital swelling and in more advanced cases, proptosis, ophthalmoplegia, and optic nerve dysfunction. Up to one-third of these cases are bilateral. Prompt diagnosis is critical, as delay may lead to permanent visual loss. Computed tomography and magnetic resonance orbital imaging are usually needed to confirm the diagnosis. Conservative treatment includes intravenous fluids, blood transfusion, and steroids with antibiotic cover in order to treat the vaso-occlusive crisis and reduce orbital inflammation.
TABLE 17-3 Clinical Characteristics of Orbital Compression Syndrome in the Literature (n= 27)
Modified from Dixit A, Chatterjee TC, Papneja M, et al. Sickle beta-thalassemia presenting as orbital compression syndrome. Ann Hematol. 2004;83:536.
Anterior Segment
Conjunctival Sickle Sign
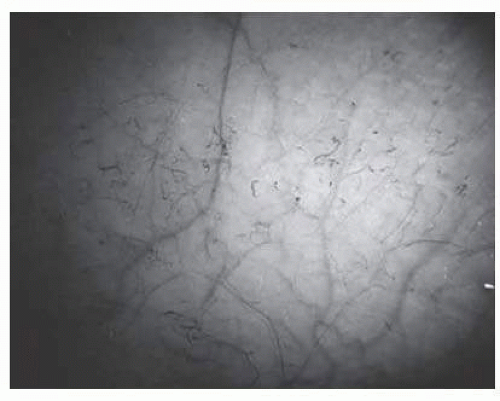
Abnormalities of the bulbar conjunctival blood vessels, one of the earliest reported ocular changes, provided direct evidence of the vaso-occlusive process in hemoglobinopathies.63, 64, 65, 66, 67 and 68 The theory that these abnormalities are the result of flow obstruction or impedance by sickled cells is supported by histologic examination of conjunctival vessels: trapping of erythrocytes in distal portions of capillaries with dilatation and thinning of proximal segments.69 The severity of the conjunctival changes ranges from linear dilatations to isolated groups of truncated, comma-shaped segments. These changes have been correlated with the ISC count, Hb S concentration, and the intraerythrocytic hemoglobin concentration (Fig. 17.1).70, 71, 72 and 73 Although known as the conjunctival sickle sign, these vascular abnormalities are not completely pathognomonic of sickle cell disease. In rare cases, they are seen in patients with AIDS, chronic myelogenous leukemia, and other vaso-occlusive diseases.73, 74 and 75 The accessibility of the conjunctival vessels has facilitated the noninvasive evaluation of potential treatments for sickle cell disease. Rodgers et al.76 demonstrated a significant decrease in the amount of blanching of the conjunctiva after the administration of the vasodilator, nifedipine, suggesting improved blood flow. In a randomized, placebo-controlled trial,77 Poloxamer 188 was shown to significantly reverse conjunctival microvascular changes caused by vaso-occlusive crises. While neither of these treatments has proven to be efficacious systemically in the treatment of patients with sickle cell disease, the ability to directly view the vaso-occlusive process in the conjunctival vessels may help to evaluate other therapies in the future.
FIG. 17.1 Conjunctival vascular abnormalities in a patient with homozygous sickle cell disease demonstrating interrupted, dilated, and truncated vascular segments.
Newer technologies are being employed to investigate the conjunctival microcirculation. In a study using Computer-assisted intravital microscopy (CAIM), abnormalities in vessel morphometry,78 vessel tortuosity, and distribution were found to be more severe in adult than pediatric patients with sickle cell disease. Abnormal flow (sludged flow and boxcar) were prevalent in both children and adults. With these newer tools, conjunctival vascular abnormalities can be quantified and may ultimately prove useful as a biomarker of disease.
Iris Atrophy and Neovascularization
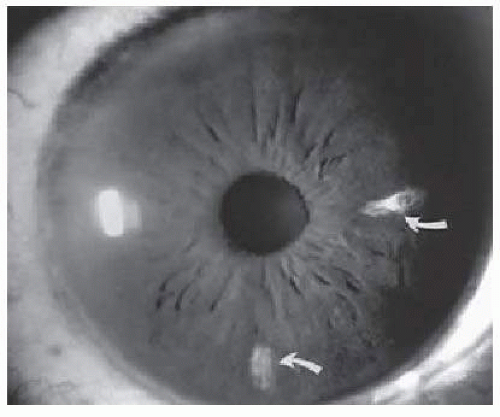
Iris atrophy may develop secondary to the occlusion of iris vessels.79, 80 The area of atrophy may be extensive (Fig. 17.2) and may be associated with pupillary irregularity. Iris atrophy may be more prevalent in Hb SC disease. Acheson et al.81 found a 14.7% prevalence rate of iris atrophy among males with Hb SC disease, with a close association with proliferative sickle retinopathy in the same eye. Iris neovascularization may develop in eyes with chronic retinal detachment or major arteriole occlusions and can, in rare cases, cause a secondary neovascular glaucoma.82
FIG. 17.2 Iris infarcts in a patient with Hb SC disease(arrows).
Hyphema
Sickle cell patients, including patients with sickle cell trait, have a higher incidence of increased intraocular pressure (IOP), optic nerve atrophy, and secondary hemorrhage in the setting of traumatic hyphema compared with non-sickle cell patients.83, 84, 85 and 86 In a study of 99 patients with hyphema, 92% of sickle cell-positive patients had an IOP greater than 22 mm Hg within 48 hours compared with only 19% of sickle cell trait-negative patients.87 The mechanism of the elevation in IOP is thought to be secondary to Hb S polymerization in the aqueous humor, causing sickling of RBCs and subsequent blockage of the outflow channels of the trabecular meshwork.84, 85, 86, 87, 88, 89, 90, 91, 92 and 93
The potential for permanent ocular damage exists in sickle cell disease patients with even minimal hyphema from surgery or anterior segment trauma, as these patients are more susceptible to developing central retinal artery occlusions and optic atrophy even at mildly elevated IOP.94, 95 Even in patients with sickle cell trait, moderately elevated IOP can cause optic atrophy in as few as 2 to 4 days.96
A sickle cell screening test is recommended for every African-American patient who presents with a hyphema and for every patient with hyphema associated with elevated IOP.97, 98 If Hb S is present, the IOP should be closely monitored and should average no higher than 24 mm Hg for more than 24 hours.85, 86 Medical management should avoid treatments that promote sickling. A topical β-blocker is a good initial therapy, followed by topical brimonidine. The use of topical dorzolamide is controversial. Systemic carbonic anhydrase inhibitors promote hemoconcentration, induce systemic acidosis, and exacerbate sickling. Acetazolamide lowers aqueous pH and potentially causes further Hb S polymerization and sickling. Methazolamide causes less systemic acidosis and may be used with caution (dose: 10 mg per kg divided three times a day to a maximum of 50 mg three times a day).97 Other osmotic agents (glycerin, isosorbide, intravenous mannitol) can also cause hemoconcentration and increased blood viscosity and, thus, should be used sparingly (every 24 hours). Oxygen (100%) inhalation or oxygen goggles, which deliver oxygen through the cornea to the aqueous humor, may also reduce sickling in the anterior chamber. Despite maximal medical management, if the average IOP is greater than 24 mm Hg over any 24-hour period and repeated transient spikes occur above 30 mm Hg, surgical intervention with anterior chamber lavage and hyphema evacuation is indicated.99 Gonioaspiration has been reported as an alternative in sickle trait patients to the anterior chamber washout procedure.100 Systemic epsilon aminocaproic acid has been evaluated and shown to be effective in preventing rebleeding in traumatic hyphemas and should be used in sickle cell patients.101
Posterior Segment
Optic Nerve: Disc Sign
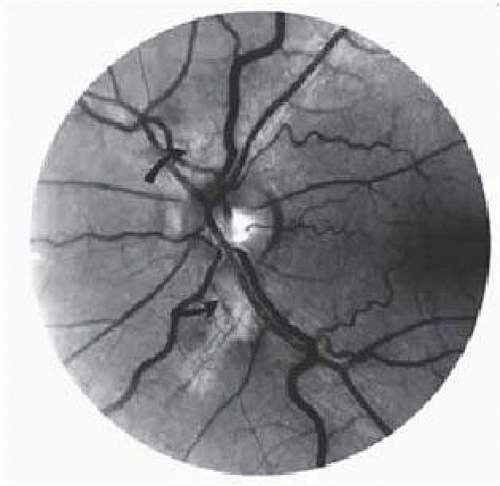
Transient dark red spots (similar to conjunctival commas), representing plugs of sickled erythrocytes within superficial capillaries, may be seen on the surface of the optic disc (Fig. 17.3, Color Plate 17.1A). These disc changes are not associated with any functional or anatomic abnormalities. They are found in 11% of all patients with sickle cell disease but appear to be more common in patients with homozygous sickle cell disease, occurring in 29% of these patients.102 The disc sign correlates with the presence of conjunctival commas and ISCs.
Unlike some retinal vaso-occlusive diseases, sickle cell retinopathy is rarely associated with optic disc neovascularization.103, 104 In our extensive experience with sickle cell disease, we have seen only one such case. The low incidence of optic disc neovascularization may be due to the peripheral location of the ischemia and to the localized changes, much of the retina not being significantly affected by the ischemia.104 If optic disc neovascularization does occur, peripheral retinal scatter photocoagulation is effective in stimulating regression.104
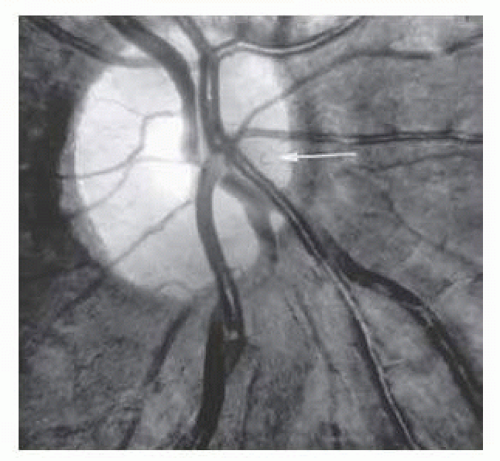
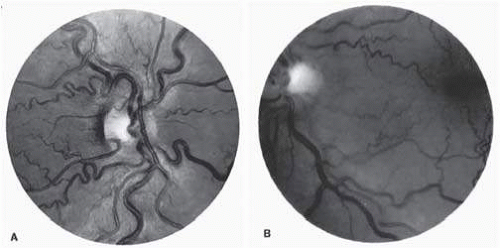
Dilation and tortuosity of the retinal veins was one of the first recognized abnormalities of sickle cell eye disease. Although it is not pathognomonic of sickle cell disease, it reportedly occurs in up to 47% of patients with homozygous sickle cell disease and 32% of patients with SC disease (Fig. 17.4).105 In a study of 70 asymptomatic patients with hemoglobinopathy, retinal vein dilation and tortuosity was observed in 54% of Hb S-β-thalassemia and 83% of Hb SS patients.106 The significance of this venous tortuosity is unknown, and the incidence does not appear to be related to age.107
Angioid Streaks
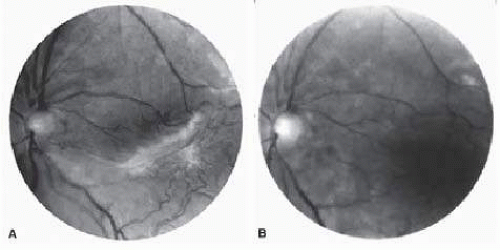
Angioid streaks occur in association with sickle cell disease, with an overall incidence of less than 6%.108, 109, 110 and 111 The changes are more common in patients with homozygous sickle cell disease and are age-dependent, occurring in 2% of sickle cell disease patients less than 40 years of age versus 22% in those who are more than 40 years of age (Fig. 17.5).112
Unlike the angioid streaks seen in patients with pseudoxanthoma elasticum (PXE), choroidal neovascularization and disciform disease are uncommonly associated with sickle cell disease. Previously, elastic tissue degeneration, as is seen in PXE, had not been demonstrated in the skin biopsy specimens of sickle hemoglobinopathy patients with angioid streaks.109, 111 However, recent evidence has suggested an underlying generalized elastic tissue defect in sickle cell disease and β-thalassemia.113 In a study of 100 patients with β-thalassemia, 16% had PXE-like cutaneous lesions confirmed by skin biopsy, and 20% had angioid streaks.114 In addition, arterial calcifications similar to PXE have been reported in thalassemia.113 Initially, the etiology of angioid streaks in sickle cell disease was hypothesized to be due to chronic hemolysis and iron deposition, causing brittleness of Bruch membrane. Histopathologic examination of angioid streaks in a patient with homozygous sickle cell disease, however, revealed heavy calcification of Bruch membrane without evidence of iron or hemosiderin.115
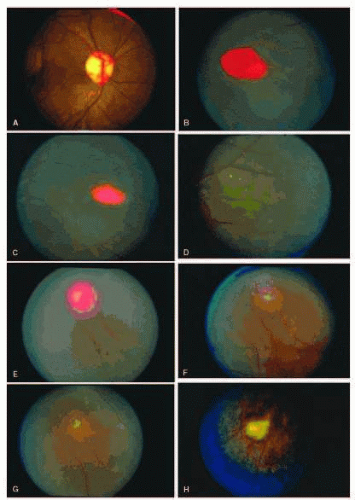
COLOR PLATE 17.1 A: Photograph of the disc in the same patient as in Figure 17.16A, demonstrating the disc sign with multiple dilated vascular segments. B: Salmon-patch hemorrhage with preretinal blood obscuring the retinal vasculature. (B-G from Gagliano DA, Goldberg MF. The evolution of salmon-patch hemorrhages in sickle cell retinopathy. Arch Ophthalmol. 1989;107:1814, with permission.) C: Two weeks later, the hemorrhage shown in B has a central grayish white color as it begins to resolve. D: Two years later, the hemorrhage shown in B and C has resolved, and an iridescent spot remains. E: Salmon-patch hemorrhage with pre-, intra-, and sub-retinal blood. F: Two months later, the hemorrhage shown in E has resolved, and a lightly pigmented area surrounded by a depigmented halo is seen. G: Four years later, a well-pigmented black sunburst adjacent to the arteriole remains from the salmon-patch hemorrhage shown in E. H: Iridescent spot with refractile copper-colored granules.
FIG.17.4 A: Generalized vascular tortuosity, predominantly venous, in a patient with homozygous sickle cell disease. B: Localized macular venous tortuosity in a patient with heterozygous SC disease.
FIG. 17.5 A 45-year-old man with homozygous sickle cell disease and angioid streaks (arrows).
Epiretinal Membranes
Epiretinal membranes may produce visual loss in patients with sickle cell disease. Macular epiretinal membranes are seen more frequently in eyes with retinal neovascularization, retinal tears, and vitreous hemorrhage, as well as in eyes that have had laser treatment or surgery of the retina or vitreous.116 Progressive visual loss from macular distortion has been reported in up to 32% of these patients over a 2.5-year period.117 Peripheral neovascularization may stimulate formation of epiretinal membranes by transudation of plasma and erythrocytes into the vitreous, disrupting the vitreous cortex and inducing posterior vitreous detachment. Cytokines associated with retinal ischemia may be released and play a role. Successful treatment of the neovascular tissue reduces the risk of epiretinal membrane development by approximately 30%.117 Although spontaneous separation of epiretinal membranes following treatment of peripheral neovascularization has been observed, surgical removal may be considered when patients exhibit moderate to severe visual loss (Fig. 17.6).118, 119 Traction across the macula from peripheral neovascularization is thought to contribute to the formation of macular holes in sickle cell retinopathy.120 Vitreous degeneration and anterior-posterior traction may also play a role. Successful surgical closure of macular holes in patients with PSR with concomitant improvement in vision is possible.121
Retinal Artery Occlusions
Occlusions of the central retinal artery and major arteriolar branches are probably most frequent in young patients with homozygous sickle cell disease; however, they may also occur with other sickling genotypes (Fig. 17.7).64, 122, 124 They may cause permanent or transient visual loss and can occur simultaneously in both eyes.126, 127, 128 and 129 There has been one reported case of partial macular sparing in a central retinal artery occlusion due to sickle cell disease.130 Arterial occlusion has also been reported to occur as a complication of retrobulbar anesthesia with131 and without retrobulbar hemorrhage,132 and following compression of the eye during photocoagulation (Fig. 17.8). Characteristic hemorrhages have also been described following central retinal artery occlusions.133
FIG. 17.6 A: Preoperative photograph of the left eye of a 64-year-old woman with SC disease, who received cryotherapy and scatter photocoagulation therapy for proliferative sickle retinopathy, resulting in the development of an epiretinal membrane. At this point, visual acuity was 20/400. B: Postoperative photograph after vitrectomy and removal of the epiretinal membrane. Visual acuity was improved to 20/60.
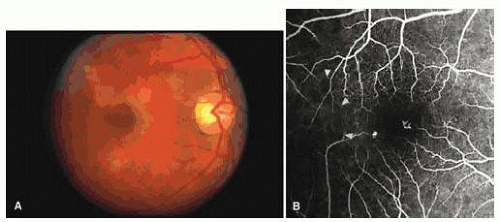
FIG. 17.7 Transient perimacular arteriolar occlusions in a 32-year-old patient with SC disease, who presented with decreased vision in the right eye (20/40) after being tackled while playing football. A: Photograph of right macula showing a white, opaque retina and a cherry red spot due to multiple arteriolar occlusions. B: Fluorescein angiogram shows multiple avascular areas, particularly at the temporal raphe (arrowheads), and an irregular perifoveal capillary network (open arrows).
While sickle cell trait was previously thought to be a relatively benign condition, retinal artery occlusion has been reported in several patients with sickle cell trait. These occlusions have been associated with airplane travel,105 elevated IOP following blunt trauma,94, 95 extreme dehydration,134 rheumatoid arthritis,135 tuberculosis, and systemic lupus erythematosus.136
Macular Small Vessel Occlusions
Occlusions of the fine vasculature of the macular and perimacular area have been reported in 10% to 40% of patients with sickle cell disease.25, 112, 137, 138, 139, 140, 141, 142, 143, 144 and 145 Macular occlusion may be the presenting sign of a hemoglobinopathy.146, 147 In the acute phase, the occluded vessel will have a dark-red appearance and may appear as a dark line on fluorescein angiography (Fig. 17.9). Nerve fiber layer infarcts (cottonwool spots) are seen (Figs. 17.8D, E and 17.10).148
Only gold members can continue reading. Log In or Register to continue