RHEUMATOLOGIC DISEASES
Rheumatologic diseases have patterns of organ involvement that not only overlap each other but overlap other diseases as well, which frequently makes specific diagnosis difficult. When first recognized as a distinct histopathologic characteristic for a collection of diseases, perivascular collagen deposition prompted the name collagen vascular diseases. However, the term “collagen vascular disease” is a misnomer, these diseases affect many different proteins in addition to the collagen, and they affect many structures in addition to vascular structures. Subsequently, the association of the collagen vascular diseases with immunologic reactions to body proteins prompted many to call these disorders systemic autoimmune diseases. This collection of diseases includes not only classical connective tissue diseases, such as systemic lupus erythematosus (SLE), Sjögren syndrome, and scleroderma, but also vasculitides and musculoskeletal autoimmune syndromes. In contrast with systemic autoimmune disease are the organ- or tissue-specific autoimmune diseases, such as type I diabetes mellitus, Hashimoto thyroiditis, myasthenia gravis, and multiple sclerosis. In addition, many pure connective tissue diseases (such as scurvy, Ehlers-Danlos, and Marfan syndrome) are not autoimmune in nature.
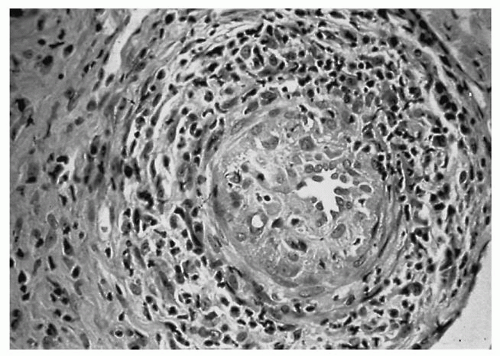
The exact cause of systemic autoimmune diseases remains obscure. The prevailing histopathologic feature of these diseases is a varying amount of connective tissue and blood vessel inflammation with abundant fibrinoid deposits (
Fig. 17.1). The tissue distribution of the inflammatory response and the pattern of organ involvement differentiate one systemic autoimmune disease from another. Specific autoantibodies are associated with some of these entities (
Table 17.1), although many of these autoantibodies are present in several autoimmune diseases and other conditions. Thus, clinical diagnosis is based on the constellation of symptoms, signs, laboratory tests, and histopathology. Diagnostic criteria have been developed for the majority of these conditions.
Systemic Lupus Erythematosus
SLE is a common systemic autoimmune disease. According to a recent report from the National Arthritis Data Working Group, approximately 250,000 Americans have systemic lupus (
1), making the incidence of the disease approximately 1 in 1,400, with nine women affected for every man. The course of SLE is highly variable, affecting primarily women of childbearing age. The disease also is three times more common in African-Americans than in white Americans. Protean systemic manifestations include arthritis, constitutional symptoms, photosensitive skin eruptions, serositis (pleurisy, pericarditis), nephritis, leukopenia, anemia, hypercoagulability, and central nervous system (CNS) involvement (most commonly cognitive dysfunction and intractable headaches). The classification
criteria for SLE are outlined in
Table 17.2 (
2). Infectious complications related to active SLE and immunosuppressive treatment are now the most common cause of death in early active SLE, and accelerated arteriosclerosis is the main cause of late mortality (
3), although the survival rate with this disease is now about 90% at 10 years and 70% at 20 years; this rate has been improved over time (
4).
Head and Neck Manifestations
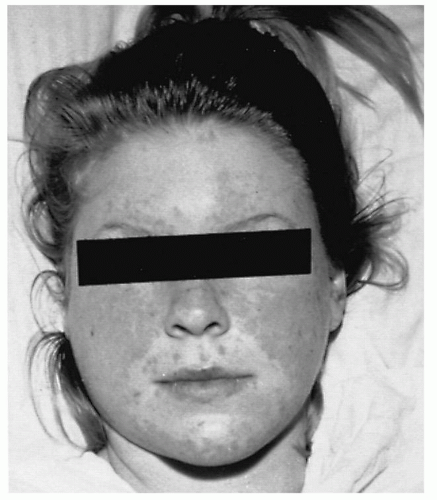
The head and neck manifestations of SLE are dominated by skin and mucosal lesions. In 35% to 40% of patients, a malar or “butterfly” rash is the first sign of the disease and is commonly precipitated by sunlight exposure (Fig. 17.2). Thirty-seven percent of patients with SLE have photosensitive skin, and sunlight exposure can trigger not only the skin rash but also systemic disease exacerbations. Additional manifestations of SLE are seen in many forms. Associated oral lesions are in general of three types: erythematous, discoid, and ulcerative. Discoid lesions are superficial; have a well-defined elevated border; may be painful; and show pronounced hyperemia, edema, and white dots with a tendency toward bleeding. Localized telangiectasia may produce a red halo effect around the affected mucosa, and secondary moniliasis and xerostomia also are common. Ulcerative lesions are usually shallow, often occur in crops, most commonly are found on the hard palate, but the buccal mucosa or nasopharynx is affected in one-third of patients. Involvement of larynx and nasal mucosa has been described. Mucosal ulceration is painful, may persist or be cyclical, and tends to occur with disease flare. Erythematous lesions are the most common; they are usually painless, red, flat, and have ill-defined borders. Histologically, mucosal lesions in SLE demonstrate orthokeratosis and parakeratosis alternating with areas of epithelial atrophy. Keratotic plugging, acanthosis, and pseudoepitheliomatous hyperplasia are common findings. Superficial, perivascular, and deep lymphocytic infiltration can be seen throughout the mucosa, and skin lesions may show only inflammation or, by immunofluorescence (IF), immune deposits at the dermal-epidermal junction.
Numerous other head and neck problems may be associated with SLE. In 3% to 5% of well-established cases,
recurrent nasal mucosa ulcerations occasionally lead to nasal septal perforation. Inflammatory changes of the larynx and trachea have been described, including true vocal fold thickening or paralysis, cricoarytenoid arthritis, hoarseness due to recurrent laryngeal nerve paralysis, edema with airway obstruction due to necrotizing vasculitis, and subglottic stenosis. Up to 25% of SLE patients report dysphagia. In 10% of SLE patients, acute enlargement of the parotid gland occurs and may be unilateral, tender, and confused with acute parotitis. Additionally, xerostomia may become a chronic problem. Sicca complex alone or in association with secondary Sjögren syndrome is a common finding in SLE patients.
Neuropathy also is a major characteristic of SLE, and in 15% of patients, the cranial nerves are affected. The neuropathy may involve the motor supply to the extraocular muscles, the sensory divisions of the trigeminal nerve, the motor divisions of the facial nerve, the vestibular portion of the vestibulocochlear nerve, or the optic nerve. Sudden hearing loss also has been described with SLE, although a definitive link has not been established. Such hearing loss may be due to thrombosis or vasculitis, although temporal bone studies have been inconclusive. A nonspecific lymphadenopathy associated with SLE in some cases appears to be related to skin or mucosal lesions.
Discoid lupus erythematosus is a subtype of SLE in which cutaneous lesions result in significant scarring but with no visceral involvement. These lesions are welldemarcated, erythematous, edematous papules that depigment and scar on resolution. The face is involved in 85% of cases, the scalp in 60%, and the ear in 44%. Associated leukoplakia of the tongue and oral mucosa may occur.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease of unknown cause that primarily presents with inflammation of synovial tissue. Symmetrical involvement of the peripheral joints is the dominant clinical feature. The course of the disease is variable, usually progressive, and may affect nonarticular tissues. RA occurs in 1% of the population, affecting women two to three times more often than men. Although it may occur at any age, and a distinct juvenile variety exists, it is more common in the fourth to fifth decade. The onset of RA may be acute, but more frequently
it is insidious, with progressive joint involvement. Morning stiffness lasting more than 30 minutes and stiffness after prolonged inactivity are common symptoms. Tenderness and inflammation in an inactive joint are physical findings specific to RA, and subcutaneous rheumatoid nodules aid in the diagnosis. Nonarticular manifestations include visceral nodules, vasculitis, pleural or pericardial effusions, and Sjögren syndrome. The classification criteria for RA are found in
Table 17.3 (
8).
Head and Neck Manifestations
Articular involvement predominates in the diverse head and neck manifestations of RA, affecting the ossicles, temporomandibular joints, cricoarytenoid joints, and the cervical spine. Temporomandibular joint dysfunction may be prominent; causing many patients with RA to have temporomandibular joint complaints including pain or tenderness at the joint or in the masseter or temporalis muscles, crepitus, limited mobility, or deviation, and radiographic evidence of joint erosion is often present. Temporomandibular joint dysfunction in patients with RA may be severe and cause contractures of the muscles of mastication, producing an anterior open-bite deformity (
9).
RA is the most frequent cause of arthritis in the cricoarytenoid joint. Histologic abnormalities of the cricoarytenoid joints are present in 86% of patients with RA. Clinically, however, only 30% of the patients with RA are hoarse. Cricoarytenoid arthritis may occur with dyspnea on exertion, anterior neck or ear pain, fullness in the throat, dysphagia, and aspiration. Hoarseness in RA is usually the result of cricoarytenoid joint involvement but may be caused by rheumatoid nodules within the cords and ischemic recurrent nerve paresis or paralysis (
10). The sudden onset of stridor and dyspnea in a patient with RA is an emergency requiring systemic steroids and possibly a tracheostomy. The oral cavity is not usually involved with abnormalities related to RA unless associated Sjögren syndrome is present or medication side effect develops. In an uncommon variant of rheumatoid vasculitis, oral ulcers similar to those seen in polyarteritis nodosa (PAN) are found.
The middle ear may be involved in severe cases of RA if synovitis develops in the ossicular joints, but this occurrence rarely results in a conductive hearing loss, except during an acute RA flare. Stiffness in the incudomalleolar and incudostapedial joints does not impair sound conduction but does result in stiffness abnormalities detected on tympanometric testing. Autoimmune inner ear disease also has been related to RA, but no definitive mechanism has been proven (
11).
Long-standing active RA may lead to cervical spine disease. Recurrent tenosynovitis involving the transverse ligament of atlas results in laxity and/or odontoid process erosion, in which case the ring of C1 can move forward in flexion causing cord compression. Any clinician should be aware about this entity and maintain a high index of suspicion when symptoms such as neck pain, stiffness, or radicular pain are present. Neck manipulation during endoscopic procedures or endotracheal intubation might precipitate atlantoaxial subluxation with subsequent cord compression. Physical examination may reveal abnormal protrusion of the axial arch on the posterior pharyngeal wall, loss of occipitocervical lordosis, and resistance to passive neck motion. Radiographic studies are necessary for the diagnosis.
Sjögren Syndrome
Sjögren syndrome is a chronic disorder characterized by immune-mediated destruction of exocrine glands, which are predominantly, but not exclusively, the lacrimal and salivary glands. Sjögren syndrome is important to be understood as a slowly progressive, SYSTEMIC autoimmune disease that affects primarily the exocrine glands. Systemic involvement is common; eventually more than three-quarters will develop one or more extraglandular manifestation. Sjögren syndrome occurs in 1% of the general population and in 10% to 15% of patients with RA. A 9:1 female preponderance is noted, and onset occurs between ages 40 and 60 years. Sjögren syndrome occurs in primary and secondary forms. The primary form is a diagnosis of exclusion. The secondary form refers to the sicca complex accompanying any of the systemic autoimmune diseases. The secondary Sjögren syndrome is seen primarily with RA but is also commonly associated with SLE, scleroderma, and primary biliary sclerosis. In one-third of patients with primary Sjögren syndrome, the disorder is systemic with involvement of extraglandular sites (arthritis 70%, Raynaud phenomenon 40% to 50%, pulmonary involvement 25% to 30%). Sjögren syndrome is associated with an increased (33 to 44 times) risk of lymphoma. The greatest risk for degeneration is seen in patients with the primary form, constant parotid swelling, splenomegaly, and lymphadenopathy, low C4 complement level and type II mixed monoclonal cryoglobulinemia. The majority of lymphomas are of mucosal-associated lymphoid tissue type.
The common clinical manifestations of Sjögren syndrome include xerophthalmia with secondary keratoconjunctivitis and xerostomia, with or without salivary gland enlargement. These manifestations are called sicca complex or sicca syndrome. A salivary gland biopsy is the best single test for Sjögren syndrome (specificity 83%, sensitivity 81%). A minor salivary gland biopsy usually demonstrates heavy lymphocyte infiltration, although a parotid biopsy may be more sensitive and specific. Rheumatoid factor and antinuclear antibodies are high in most Sjögren syndrome patients. Antibodies directed toward Sjögren syndrome A (Ro/SS-A) and Sjögren syndrome B (La/SS-B) antigens are noted in 60% and 30% of patients, respectively. These antibody tests lack specificity because they are commonly found in patients with SLE, RA, and polymyositis. Anti-La Ab is more specific than anti-Ro Ab, but less sensitive. Finding anti-La alone is atypical and was found more common in primary biliary cirrhosis or autoimmune hepatitis (
12). In many anti-Ro-positive Sjögren syndrome patients, SLE will later develop. The classification criteria for Sjögren syndrome are outlined in
Table 17.4 (
13).
Confusion often is found between the designations of Sjögren syndrome and Mikulicz disease. The latter is swelling of the salivary glands accompanying nonsystemic autoimmune diseases, such as hyperlipoproteinemia, malnutrition, diabetes, cirrhosis, tuberculosis, and sarcoidosis. In the recent years, Mikulicz syndrome has been increasingly recognized as part of the spectrum of IgG4-related disease and not confined to salivary glands. It is characterized
by infiltration of IgG4-positive plasma cells in the salivary glands tissue, raised serum levels of IgG4 and fibrosis or sclerosis. Sjögren syndrome is not related to the divergent group of systemic diseases associated with Mikulicz disease.
The autoimmune diseases of the parotid gland have a unifying histologic pattern of an early lymphocytic infiltrate, followed by thinning and fragmenting of the connective tissue in the terminal or intercalated duct walls with destruction of the acini. The larger ducts are usually uninvolved unless a superimposed infection is present. A diffuse pattern of globular collections of contrast material is observed, originally called sialectasis. It was initially thought that dilatation of the acini and collection of the contrast material within them was noted. It is now known, however, that the weakened acini allow extravasation of contrast material, which forms extramural globular collections. The four progressive radiographic features of autoimmune parotid swelling are punctate, with spherical collections of 1 mm or less; globular, with collections of 1 to 2 mm; cavitary, with large irregular collections of uneven distribution; and destructive, with no recognizable branching. The latter two stages represent superimposed infection.
Head and Neck Manifestations
Exocrine gland pathology dominates the head and neck manifestations of Sjögren syndrome. About 80% of these patients complain of xerostomia, the most prominent symptom of the disease. These patients report difficulty chewing, dysphagia, dysgeusia, fissures of the tongue and lips, and an increased number of dental caries. Oral candidiasis and angular cheilitis are frequent complications of dry mouth. Additionally, diminished secretion of tears leads to keratoconjunctivitis sicca and eye complaints, including dryness, burning, itching, and foreign-body sensation. Loss of glandular secretions in the nasal passages causes crusting and secondary epistaxis in 50% of patients and hyposmia in 40%. Chronic sinusitis may result from inspissated secretions, and occlusion of the nasolacrimal ducts may occur. Other manifestations of primary Sjögren syndrome include sensorineural hearing loss and, in 30% of patients, a painless intermittent unilateral parotid swelling of unpredictable duration that is rarely associated with edema.
Systemic Sclerosis (Scleroderma)
Scleroderma, also called systemic sclerosis (SSc), is characterized by sclerotic skin changes and is often accompanied by a multisystem disease. Progressive fibrosis of involved organs is the pathologic hallmark of the disease. Scleroderma is usually divided into two main forms: localized scleroderma and SSc. This division has important prognostic implications and allows specific clinical approach. SSc is further divided into limited cutaneous disease (lcSSc) and diffuse cutaneous disease (dcSSc). LcSSc is characterized by skin thickening distal to the elbows and knee, severe Raynaud phenomenon preceding skin disease, anti-centromere antibodies, and later development of visceral involvement (gastrointestinal and lung, pulmonary artery hypertension [PAH] and interstitial lung disease). DcSSc is characterized by proximal skin thickening (thighs, trunk, and arms), significant visceral disease (lung, heart, kidney, and gastrointestinal), absence of anticentromere antibodies, and a worse prognosis (
15). The incidence is approximately 18 to 20 new cases per million per year and the disease has a 3:1 female-to-male preponderance, with 50 years as the median age of onset. Initial presentation includes Raynaud phenomenon, edema of the fingers and hands, and skin thickening. The American College of Rheumatology criteria for scleroderma include one major criterion (sclerodermatous skin changes proximal to the metacarpal-phalangeal joints) and two of three minor criteria: sclerodactyly, evidence of ischemia of the digits (digital pitting scars), and bibasilar pulmonary fibrosis on chest radiograph (
16). These criteria were found to have 97% sensitivity and 98% specificity for the diagnosis of scleroderma. Visceral and often fatal manifestations are seen in the gastrointestinal tract, lung, heart, and kidneys. Arthralgias and muscle weakness are common musculoskeletal complaints, and Raynaud phenomenon is almost universal.
Head and Neck Manifestations
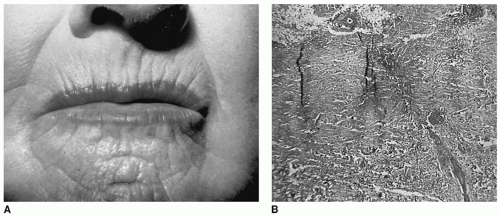
Eighty percent of patients with SSc have signs and symptoms involving the head and neck, and in 30% of those patients, the head and neck symptoms are part of the presenting complaints. In the skin, edema precedes epidermal atrophy and loss of appendages. Eventually, in 35% of these patients, facial tightness develops. A decreased ability to open the mouth is the initial complaint in 19% of patients, which is a secondary manifestation to skin changes. Additional skin manifestations include telangiectasias (19%), calcinosis (3%), and linear scleroderma, which usually affect the scalp and limbs. The typical facies associated with scleroderma consists of tight skin, thin lips, and vertical perioral furrows (
Fig. 17.3). These skin changes represent a major aesthetic concern for patients (
17). These
skin changes are secondary to the underlying dermal and subcutaneous inflammatory process. Dysphagia is the most common initial complaint. Esophageal problems occur in nearly all patients with this disease; abnormal radiographic findings are observed for the distal two-thirds of the esophagus in 80% of patients with SSc, and 50% of those patients are symptomatic. The dysphagia is due to involvement of esophageal innervation, smooth muscle (the distal two-thirds of esophagus, the portion composed of smooth muscle), esophageal stricture, or combination of all. Upper esophageal involvement does not usually occur unless there is an overlap with inflammatory myopathy, in which case even pharyngeal involvement can be seen, because of the striated muscle structure in the proximal esophagus. Decreased or absent peristalsis in the distal esophagus with mild to moderate dilation also is reported, and hiatal hernias are common.
Scleroderma often involves structures in the head and neck. Gingivitis and periodontal membrane thickening are common. Oral tissues demonstrate edema followed by atrophy and induration of mucosal and muscular tissues. About 25% of patients report xerostomia, xerophthalmia, or both (the sicca complex). A minor salivary gland biopsy will not demonstrate the lymphocytic infiltrate seen with Sjögren syndrome but will demonstrate fibrosis. Involvement of the larynx also occurs, and almost half of the patients with SSc complain of a voice change. The most common cause for laryngeal disease in scleroderma is chronic acid irritation from severe esophageal disease and incompetent lower esophageal sphincter. Raynaud phenomenon of the tongue, an unusual complication, may appear as mucosal blanching associated with dysarthria. Trigeminal neuralgia and facial nerve palsy are infrequent manifestations. Patients with SSc, particularly dcSSc, are at increased risk for development of carcinoma of the tongue (
18).
Inflammatory Muscle Disease (Polymyositis and Dermatomyositis)
The inflammatory myopathies are a group of disorders characterized by proximal muscle weakness and nonsuppurative inflammation of the skeletal muscle. Polymyositis, dermatomyositis, and inclusion body myositis are subsets of this group. The incidence of these disorders is estimated to be approximately five new cases annually per million, with a 2:1 female preponderance, and the age of onset is between 40 and 50 years, with a pediatric variant affecting children between the ages of 5 and 15 years. The diagnostic criteria for polymyositis and dermatomyositis are complex and subject to ongoing debate (
21,
22,
23) (
Table 17.5). The most common presentation is that of progressive proximal muscle weakness over a period of months, and fatigue is common.
The inflammatory myopathies may be isolated or associated with other abnormalities. Polymyositis and dermatomyositis may be associated with other systemic autoimmune disorders, including SSc, SLE, Sjögren syndrome, Churg-Strauss syndrome, and RA. In approximately 20% of patients, the myopathy is associated with a malignancy, particularly ovarian cancer, but non-Hodgkin lymphoma and carcinoma of the lung, prostate, breast, nasopharyngeal areas, and colon have also been implicated.
Head and Neck Manifestations
Head and neck manifestations of polymyositis reflect proximal muscle involvement. Half of the patients report weakness of the neck muscles, which is often manifested by a patient’s inability to lift his or her head off a pillow. Difficulty in phonation and deglutition occurs because of diseased tongue muscles, and nasal regurgitation is common because of palatal and pharyngeal muscle involvement. Thirty percent of patients with these diseases have upper dysphagia secondary to the involvement of the upper esophagus, pharynx, superior constrictors or, most commonly in inclusion body myositis, the cricopharyngeus (
24). Dysfunction of these muscle groups also results in aspiration and secondary pneumonia. Cutaneous photosensitivity is common in dermatomyositis. The heliotropic rash, seen in some (fewer than 50%) patients with dermatomyositis, is a purplish discoloration in the periorbital area, especially the upper eyelids.
Relapsing Polychondritis
Relapsing polychondritis is characterized by episodic recurring inflammation of the cartilaginous structures and other tissues with a high concentration of glycosaminoglycans that are eventually replaced by granulation tissue and fibrosis. Diagnosis is based on chondritis at two of three sites (auricular, nasal, or laryngotracheal) or one of those three sites with two of the following symptoms: ocular inflammation, cochlear or vestibular damage (or both), or seronegative inflammatory arthritis (
27). Biopsy of the lesion is not required for diagnosis. The incidence is about 3.5 cases per million.
Head and Neck Manifestations
Auricular chondritis and nonerosive arthritis are the most common presenting symptoms of relapsing polychondritis. Auricular chondritis is characterized by the sudden onset of erythema and pain, sparing the lobule, which lacks cartilage. Chondritis is the feature presentation in 33% of patients and will develop in 90% of those with the disease. Resolution can occur in 5 to 10 days with or without treatment. Conductive hearing loss may develop secondary to collapse of the external auditory canal or eustachian tube chondritis, causing serous otitis media, and 40% have cochlear or vestibular dysfunction, possibly due to vasculitis of the internal auditory artery. Aphthous ulcers are the most common dermatologic manifestations of this disease.
Chondritis of the nasal cartilages develops in 61% of patients and often does not coincide with auricular involvement (
27). The nasal cartilage chondritis also has a sudden onset and resolves in several days with or without treatment. Overtime, and with repeated attacks, deformities result from the loss of cartilage. These deformities occur in the pinna, which become soft and flops over, and in the nasal bridge, which can cause a classic saddle deformity of the nose.
Laryngeal involvement is seen early with a nonproductive cough, which progresses to hoarseness and stridor. On physical examination, tenderness over the thyroid cartilage and the anterior trachea can be observed. Inflammation of the tracheobronchial tree damages the cartilaginous rings, which can cause a dynamic obstruction in inspiration. Stricture can form in subglottic space, increasing susceptibility to secondary infection. Of patients with relapsing polychondritis, 53% will have respiratory tract involvement during the course of their disease. Diagnostic endoscopy can be dangerous in these patients because of the risk of tracheal collapse. With extensive airway involvement, management is difficult, even with a tracheotomy. Indications for the need of a tracheotomy include severe edema of the glottic and subglottic regions of the larynx and laryngeal collapse.
Biopsy often reveals a lack of basophilic staining of the cartilage, perichondrial inflammation, with presence of mononuclear cell infiltrate at the fibrochondrial junction, and eventual cartilage destruction and replacement with fibrous tissue. The erythrocyte sedimentation rate (ESR) is often increased with active disease. Serologic evaluation often reveals a positive rheumatoid factor, antinuclear antibody, and anti-type II collagen antibody; however, the first two lack specificity and the last one is not routinely available. The clinical course ranges from mild disease to severe fulminating attacks. Mortality is generally related to respiratory involvement (i.e., laryngeal collapse) or cardiovascular disease (e.g., aneurysms or valvular insufficiency). The cause of relapsing polychondritis is unknown.
Mixed Connective Tissue Disease
The term
mixed connective tissue disease (MCTD) was coined in 1972 to describe a distinct entity with coexisting features of SLE, SSc, and myositis. This entity is characterized by high titers of anti-U1 RNP (1:1,600 or higher), a ribonucleoprotein antibody. The prevalence is unknown, and no consensus diagnostic criteria have been developed; the nature and existence of the disease as a unique entity is still debated. Eighty percent of patients are women, and onset usually occurs between the ages of 30 and 60 years, with death resulting primarily from pulmonary fibrosis and hypertension. The most acceptable criteria for diagnosis are those proposed by Alarcon-Segovia (
30). Requirements for the diagnosis are anti-U1 RNP antibody positive at titer 1:1,600 or higher plus myositis or synovitis plus two of the following: edema of the hands, acrosclerosis, or Raynaud phenomenon.
Head and Neck Manifestations
Head and neck manifestations are a combination of the features seen in other systemic autoimmune disorders. Mucocutaneous changes include malar rash, discoid lupus, sclerodermatous skin thickening, oral mucosal ulceration, and nasal septal perforation. Sjögren syndrome also has been described in 50% of patients with MCTD, albeit sicca complex is usually less prominent than in those with anti-La antibodies. Esophageal dysfunction is present in most cases, resulting in abnormal peristalsis, heartburn, and dysphagia in 60%, 48%, and 38% of patients, respectively. One interesting clinical manifestation of MCTD that happens on a fairly frequent basis, in about 25% of cases, is trigeminal neuralgia. As with most other systemic autoimmune diseases, corticosteroid and immunosuppressive agents are the mainstays of treatment with the goal of managing the specific organ involvement or disease manifestations on a case-by-case basis.
Vasculitides
The vasculitides are a group of diseases characterized by a noninfectious necrotizing vasculitis and resultant ischemia. Considerable overlap in the clinical manifestations of these diseases makes it difficult to develop categories with strict criteria. A practical approach has been to classify them into groups by the size of vessels involved, the specific anatomic sites involved, clinical manifestations, and the presence or absence of anti-neutrophil cytoplasmic antibodies (ANCAs). Some of the more important categories are discussed herein.
Polyarteritis Nodosa
PAN has been considered the prototype of the vasculitides. This rare disease has an incidence of less than 1 in 100,000 per year. It affects males and females equally and is seen in all racial groups. Patients are usually in the fifth and sixth
decades of life at presentation. PAN is a non-ANCA vasculitis affecting predominantly medium-sized arteries and can be the result of hepatitis B infection. Tissues involved include the gastrointestinal tract, hepatobiliary system, kidneys (vascular nephropathy without glomerulonephritis), pancreas, skin, testicles, peripheral nerves, and skeletal muscles. Symptoms at presentation are primarily constitutional (fever, weight loss, and malaise) with peripheral neuropathy (mononeuritis multiplex).
Despite widespread arterial involvement, otolaryngologic manifestations are few, but sudden bilateral sensorineural hearing loss has been attributed to this disease. Vestibular disturbances also have been reported. The proposed mechanism of cochleovestibular damage is thromboembolic occlusion of the end arteries of the inner ear. Other head and neck manifestations include cranial nerve palsies, in which the seventh nerve is most commonly involved, and skin or mucosal lesions.
Churg-Strauss Syndrome
Churg-Strauss syndrome, also called allergic angiitis granulomatosis, is a disease characterized by the triad of systemic small-vessel vasculitis, asthma, and hypereosinophilia. Mean age at onset is 50 years, with a male preponderance and an incidence rate of approximately three per million. Three phases of this disorder exist. The prodromal phase consists of asthma, atopy, and allergic rhinitis; the latter symptom is present in 70% of cases. The second phase is marked by hypereosinophilia and eosinophilic tissue infiltration, leading to peripheral neuropathy or pulmonary infiltrates. Systemic necrotizing vasculitis constitutes the final phase, including renal, gastrointestinal, cardiac, and CNS involvement. Progression from asthma to vasculitis occurs over a period of 3 years to decades, and diagnosis of Churg-Strauss syndrome is suggested if four of six criteria (asthma; eosinophilia greater than 10%; neuropathy, mononeuritis multiplex, or polyneuritis; non-fixed pulmonary infiltrates; paranasal sinus abnormality; and extravascular eosinophilia) are present. Seventy percent of patients have nasal involvement, with polyps, resultant obstruction, rhinorrhea, secondary rhinosinusitis, and crusting. Asthma is nearly universal, and the ANCA serology (usually perinuclear or p-ANCA detected by IF and myeloperoxidase (MPO)-ANCA detected by enzyme-linked immunosorbent assay [ELISA]) is detected in 50% of the cases. Otologic manifestations of Churg-Strauss syndrome include sensorineural hearing loss and conductive hearing loss due to eosinophilic infiltration into the mastoid and middle ear with otorrhea. The middle ear and mastoid disease is reversed by steroid treatment (
31). High-dose corticosteroids are the treatment of choice, but some patients with severe systemic involvement require cyclophosphamide. Deaths occur most commonly from cardiac and CNS involvement. Before the use of steroids, the mortality rate was 50% within 3 months of onset of the systemic vasculitis.
Granulomatosis with Polyangiitis (Wegener Granulomatosis)
Granulomatosis with polyangiitis (GPA) which is new nomenclature for the disease previously known as Wegener granulomatosis, is a rare (incidence of less than 1 in 100,000 per year) form of vasculitis characterized by the triad of upper and lower respiratory tract granulomas, vasculitis, and glomerulonephritis (Wegener triad), which could have been included in the granulomatous disease section of this chapter. No gender predilection is seen. Most patients are white and present in the fifth decade of life. The typical clinical features include bilateral pneumonitis (95% of patients), chronic sinusitis (90%), mucosal ulceration of nasopharynx (75%), and evidence of renal disease (80%). Nasal symptoms usually occur early in the disease and include crusting, epistaxis, and rhinorrhea. Erosion of the septal cartilage with saddle-nose deformity and nasal stenosis may occur after cartilage is destroyed by the vasculitis and granulomas. The sinusitis associated with GPA is difficult to treat. Staphylococcus aureus is often cultured from these patients.
The most common oral cavity findings of GPA include hyperplasia of the gingiva and gingivitis (strawberry gums). Edema and ulceration of the larynx are seen in 25% of patients. Significant subglottic stenosis develops in up to 23% of patients and is a poor prognostic sign. Salivary glands also may be involved.
Otologic problems develop in 20% to 25% of patients and include conductive hearing loss secondary to serous otitis media; suppurative otitis media, with or without granulation tissue in the middle ear; sensorineural hearing loss, often profound and bilateral; and pinna changes similar to those seen with polychondritis. Facial nerve palsies also have been documented, and is believed to be secondary to compression due to inflammation in the middle ear.
Diagnosis is based on clinical, pathologic, and laboratory findings. The hallmark pathologic lesion in GPA is necrotizing, granulomatous, small and medium vessel vasculitis with an inflammatory infiltrate. Often, upperairway biopsies are nondiagnostic because the inflammatory infiltrate obscures the vasculitis, so that multiple upper-airway biopsies or lower-respiratory biopsies are required before histologic diagnosis is ultimately made. The discovery of cytoplasmic staining ANCA (c-ANCA) has revolutionized the diagnosis of GPA. The sensitivity of c-ANCA for GPA ranges from 65% to 96% for patients with an active, generalized disease, and the specificity is quite high, although this test may be positive in patients with microscopic polyangiitis (MPA) and Churg-Strauss Syndrome. ELISA provides target antigen-specific characterization of ANCA and enhances the specificity and sensitivity of IF. The c-ANCA pattern usually corresponds to detection of proteinase-3 ANCA (PR3-ANCA) by ELISA. Finding c-ANCA by IF in combination with PR3-ANCA by ELISA carries a sensitivities and specificities >95% for GPA
(
32). Tissue diagnosis should be reserved for cases with high clinical suspicion but a negative PR3-ANCA or low clinical suspicion but positive PR3-ANCA. Classical clinical findings in conjunction with positive PR3-ANCA may be sufficient for diagnosis. The ESR varies with disease activity. Left untreated, generalized GPA is fatal within 2 years in 93% of all cases. However, symptomatic control of the disease is achieved in most patients with corticosteroids and low-dose daily cyclophosphamide (2 mg/kg/day). Some experts prefer regimens of monthly intravenous (IV) cyclophosphamide. Induction of remission with monthly IV cyclophosphamide was shown to be as effective as daily oral cyclophosphamide with fewer side effects (
33). Azathioprine or methotrexate may be alternatives to cyclophosphamide and are used for the maintenance of remission or for patients with limited disease. Recently, the RAVE study has shown that rituximab therapy was not inferior to daily cyclophosphamide treatment for induction of remission in severe ANCA-associated vasculitis and may be a better option for recurrent disease. (
34). Approximately 20% of patients, even with therapy, eventually succumb to the disease or to the therapy. Isolated sinus disease may be treated with low-dose steroids, topical nasal steroids, saline irrigations, or antibiotics when bacterial superinfection is suspected. Additionally, airway compromise may be alleviated with systemic steroids, although significant subglottic stenosis may require a tracheostomy.
Microscopic Polyangiitis
MPA is fairly recently described vasculitis, coined in 1994 by a group of experts during an international consensus conference. MPA essentially branched out from PAN as a small vessel disease. Vasculitis in the arterioles, capillaries, and venules, which involves primarily the lung and the kidney, are characteristic to MPA and absent in PAN. In addition, MPA patients commonly are ANCA positive unlike PAN. GPA, MPA, and Churg-Strauss syndrome comprise a category of small vessel vasculitis related to ANCA and are characterized by pulmonary and renal involvement. MPA and GPA have a similar clinical spectrum and are often hard to distinguish. Histopathologically, MPA is characterized by pauci-immune, necrotizing, small vessel vasculitis without evidence of granulomatous inflammation, in contrast with GPA where granulomatous vasculitis is the hallmark of disease. The prevalence is about 2 cases per 100,000 with white males being slightly more commonly affected than females; usual onset of disease is around 50 years.
The clinical presentation is similar with GPA. One of the most important differentiating factors between GPA and MPA is the severity and timing of upper respiratory tract involvement. MPA may affect the upper respiratory tract but is usually limited to a mild sinusitis, rhinitis, and mucosal lesions, and does not typically precede the disease onset. Fever, malaise, fatigue, myalgia, and weight loss is found in about 50%. Pulmonary involvement is common with infiltrates, hemoptysis, dyspnea, and cough. Renal disease in MPA is similar with GPA, but tends to present earlier in the disease. The most dreadful clinical presentation is with rapid progressive glomerulonephritis and kidney failure. Other organs and systems involvement are frequently found; patients with MPA might have gastrointestinal tract vasculitis, nervous system manifestations, more commonly mononeuritis multiplex (57%) but CNS involvement with seizure may be found in 11% of patients. Skin involvement (palpable purpura 40%, livedo reticularis, skin ulceration, and necrosis), ocular manifestation, myalgia, and arthralgia are part of the MPA clinical spectrum. Laboratory studies reveal elevated acute-phase reactants, anemia, and renal involvement (elevated BUN and creatinine, proteinuria, hematuria, and urinary casts). Eighty percent of the patients are ANCA positive and, in contrast with GPA, MPA typically exhibit a p-ANCA pattern on IF with MPOANCA reaction by ELISA. GPA typically has c-ANCA pattern on IF with PR3-ANCA by ELISA.
Treatment is identical for MPA as for GPA. The prognosis is relatively similar and the disease course is slightly different with a higher relapse rate for GPA (40%) and more frequent renal morbidity for MPA. In a retrospective study of 246 patients, 18% mortality was observed at 1 year and 76% survival at 5 years (
35).