Rhabdomyosarcoma
Geoffrey E. Bradford
David A. Kostick
First reported by Weber1 in 1854, rhabdomyosarcoma (RMS) is a heterogeneous group of soft tissue malignancies that arise from undifferentiated or pluripotential mesenchymal cells having the potential to develop into striated skeletal muscle. RMS accounts for about 3% to 8% of childhood malignancies2,3,4,5,6,7 and 20% of all malignant soft tissue tumors younger than 19 years of age in the United States2; 67% of cases involve children younger than age 10 years.8 RMS is the most prevalent extraocular orbital malignancy in children, although it is only one tenth as common as retinoblastoma.9 About 35% to 40% of RMS arises in the head and neck, and primary orbital RMS constitutes about 10% of all RMS.7,10,11,12,13 Given its undifferentiated mesenchymal cell origin, primary RMS can occur in sites where there is no skeletal muscle. Thus, in addition to primary orbital lesions, extension from RMS originating in parameningeal sites—the nasopharynx, pterygopalatine fossa, infratemporal fossa, or paranasal sinuses—or from hematogenous metastatic spread, also occurs.10,11,12,13,14
Successful treatment of RMS is among the significant advances in modern medicine. Improved outcome in patients with RMS is attributed to the emergence of radiation and systemic chemotherapy as the mainstay of treatment, a better understanding of the clinical presentation, and improved radiologic and pathologic diagnostic abilities. In the 1950s and 1960s, surgery was considered the treatment of choice, and orbital exenteration was indicated if the malignancy occurred around the eye.12 No effective chemotherapy was available, and radiation therapy was seldom used because of the fear of radiation damage to the eye and its uncertain effect on children. The overall cure rate remained low, ranging from 16% to 25%.12 In 1956, Lederman15 recommended radiotherapy for orbital tumors in general, but it was not until the report of Cassady et al16 in 1968 that radiation therapy for orbital RMS was accepted. In their report, five of five patients remained disease free for up to 4.5 years after radiation therapy for biopsy-proven orbital RMS. The addition of chemotherapy to radiation treatment was popularized after a report from the Children’s Cancer Study Group.17 This prospective, randomized study of patients with RMS who were younger than 21 years of age showed a significant improvement in survival rates with the addition of chemotherapy to the treatment regimen. The 3-year survival rate for patients with orbital RMS improved from 66% (four of six patients) treated with radiation only to 91% (10 of 11 patients) treated with radiation plus chemotherapy.18 Currently, the 5-year survival rate for patients with orbital RMS is greater than 95%.10,19,20 This chapter will review the clinical features and diagnostic evaluation of RMS, as well as provide an overview of current treatment regimens. Other recent reviews on this subject have been conducted by Shields and Shields,7 Karcioglu et al,21 Conneely and Mafee,22 and most recently by Chung et al9 and are also excellent resources for the reader.
Epidemiology
RMS has a global distribution and affects all races, with an incidence of 4.3 cases per million population per year.3 Although it may occur anywhere in the body, there are distinct clinical patterns according to the age at presentation, the histologic subtype, and the site of the tumor.23 In children it is most commonly found in the head, neck, and genitourinary system and is usually the embryonal histologic subtype. In adults, RMS typically occurs in the trunk and extremities and is often the alveolar subtype.
There is a predilection for RMS to occur in children in the first decade of life although it has been reported in patients as early as in the prenatal period24,25,26,27,28,29,30 and infancy and as late as age 68.12,31,32,33,34,35,36,37,38 The mean age at diagnosis is 6 to 8 years.7,31,32 About 16% of patients are younger than 3 years of age. There is a second, smaller peak during adolescence.3 Boys are affected more commonly than girls in a ratio of 3:2.20,32,39
Etiology
Most cases of RMS occur sporadically and their cause is largely unknown. However, in a few cases (<5%) RMS is thought to have a genetic predisposition. RMS has been associated with a cancer family syndrome—Li-Fraumeni syndrome—with a suspected inherited autosomal dominant mutation in the p53 tumor suppressor gene on chromosome 17p13.40,41 The syndrome is characterized by a high incidence of sarcomas, leukemia, brain or adrenal tumors, and premenopausal breast cancer. Moreover, Fleischmann et al42 have shown that inactivation of the known oncogene Fos in p53 mutant knockout mice leads to the formation of sarcomas that morphologically resemble the embryonal variant of human RMS.42,43 RMS has also been associated with neurofibromatosis (an abnormality also located on chromosome 17).44,45,46 Mutations on other chromosomes have also been implicated in the etiology of RMS,47,48,49,50,51,52 as well as links to environmental factors, such as maternal illicit drug use, paternal tobacco use, and lower socioeconomic status.53,54,55
Pathophysiology
Gross findings in RMS usually demonstrate a circumscribed soft, fleshy, gray to pink or yellow mass. Although grossly it may appear encapsulated, microscopically RMS has no capsule.
Three subtypes of RMS are recognized, based on histopathologic, clinical, and behavioral characteristics: embryonal, alveolar, and pleomorphic. Individual tumors may contain a mixture of two subtypes.9 The embryonal variant is the most common form found in young children (60%–90% of all cases) and is the most common form to invade the orbit (84% of all cases).56 The alveolar form is less prevalent overall (15%–20% of all cases) and is seen more in adolescents. It is also less prevalent in the orbit, constituting about 10% of all orbital RMS tumors.57,58 The pleomorphic type is rare in children and extremely rare in the orbit.6,56
The embryonal subtype typically occurs in the superonasal quadrant.4,10,19,20,32,59,60,61 A variant of the embryonal subtype is botryoid RMS, so-named because it resembles a cluster of grapes and usually occurs in infants and toddlers in the bladder or vagina, but may also arise under other mucosal membranes, such as the conjunctiva or from the paranasal sinuses.8,32,60,62 The histologic appearance of embryonal RMS is characterized by fascicles of spindle cells (strap cells) oriented in different directions within a loose, myxoid stroma (Fig. 43.1).
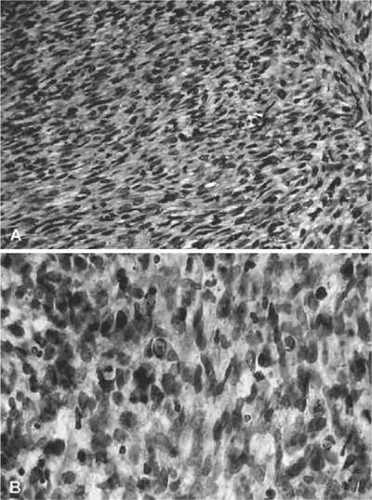 Figure 43.1. A. Rhabdomyosarcoma with spindle-shaped cells with a dense, interlacing pattern. (H&E, × 160.) B. Same specimen at a higher magnification. Note variation in size and shape of the spindle-shaped cells with a high density of nuclear chromatin. (H&E, × 640.) (Courtesy of R. Jean Campbell, MB, ChB, Mayo Clinic, Rochester, Minnesota.) |
The cells frequently contain centrally located, hyperchromatic, often bipolar nuclei (duanes fig 3a,b). Genetically, the embryonal variant is characterized by a loss of heterozygosity at the 11p15.5 locus, the region of the IGFII gene.63 Embryonal RMS is usually diploid or hyperdiploid (1.4 to 1.8).6 With treatment, embryonal RMS of the orbit carries a favorable prognosis with an overall 5-year survival rate of 66% and a 5-year survival rate of about 95% for tumors within the orbit.5,56,64
The alveolar form of RMS occurs with various histologic patterns and most often arises in the extremities, trunk, or pelvis. When present in the orbit, the inferior orbital quadrant is most frequently involved.6,12,32,60,61,65 It predominantly contains tumor cells loosely adherent to a network of thin interstitial fibrovascular septa. The tumor cells are noted to be loosely attached to the connective tissue and may become freely floating in the alveolar spaces (Fig. 43.2) mimicking, as its name implies, the histologic appearance of lung tissue.6
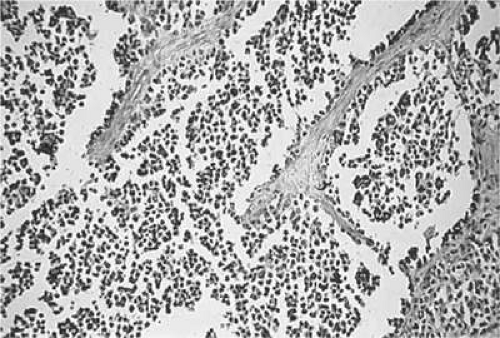 Figure 43.2. Alveolar pattern of rhabdomyosarcoma. (H&E, × 90.) (Courtesy of R. Jean Campbell, MB, ChB, Mayo Clinic, Rochester, Minnesota.) |
Compared to the embryonal variant, the alveolar subtype carries a worse prognosis: It has an overall 5-year survival rate of 54%71 and of approximately 74% for tumors within the orbit. (5,56,64) Genetically, the alveolar form (unlike the embryonal form) has consistent translocations involving t(2;13)(q35;q14) and t(1;13)(p36;q14).66 These translocations can involve the PAX3 and FKHR gene loci, located between the long arms of chromosomes 2 and 13. PCR amplification or fluorescent in situ hybridization (FISH) techniques may demonstrate these transcripts. PAX3-FKHR and PAX7-FKHR gene fusions may have prognostic implications in patients with metastatic RMS.68 Patients with alveolar RMS who exhibit the PAX3-FKHR gene fusion have a worse prognosis whereas those with the PAX7-FKHR gene fusion have a better prognosis. Gene fusion–negative patients appear to have an intermediate prognosis when compared to the gene fusion–positive groups. The alveolar variant is tetraploid (X2).6
Clinical Presentation
Orbital RMS is uniformly unilateral.9 It classically presents as a rapidly expanding mass with proptosis or displacement of the globe. (Fig. 43.3)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


