Retinoblastoma
John D. Roarty
HISTORY
Retinoblastoma was first described in 1765.1 Virchow2 identified the tumor as a “glioma of the retina” in 1864. In 1926, Verhoeff3 proposed a possible embryonic retinal origin.
Enucleation of the eye to save life was first proposed in 1809 by Wardrop.4 With improved surgical and anesthetic technique, enucleation is now the most common treatment modality.5 In 1903, Hilgartner6 successfully treated a retinoblastoma tumor with radiation, but this resulted in severe secondary ocular damage. Successful therapy with radiation followed with the refinements of Moore7 in the 1930s and Reese and colleagues8 and Stallard9 in the 1940s. In 1933, Moore successfully used interstitial radiation.
In 1953, Kupfer10 was the first to use chemotherapy to treat metastatic retinoblastoma. In the same year, Weve11 and Melchers12 reported on the use of diathermy. Because of the risk of penetration and dissemination, diathermy currently is not recommended.13 In 1959, Meyer-Schwickerwath14 reported on the use of xenon arc photocoagulation for retinoblastoma therapy.
EPIDEMIOLOGY
Retinoblastoma is the most common primary intraocular tumor in the pediatric age group.15 It represents 1%16 to 3%17 of all pediatric tumors. It is derived from retinal precursor cells.18 The incidence in the general population of the United States is in the range of 1 per 15,000 to 20,000 live births.19,20 In other countries, the range is 1 per 14,000 to 34,000 live births.15 The incidence in the United States is 10.6 per million between birth and 4 years of age, decreasing to 0.27 per million at 10 to 14 years of age.21 By 5 years of age, 90% of tumors have occurred; few adult cases are reported.15,18 Prenatal diagnosis has been achieved via ultrasound22 and amniotic genetic testing.23 No predilection based on sex, race, paternal age, or seasonal variation has been reported.24,25,26 Prenatal exposure to diagnostic irradiation does not increase the risk of retinoblastoma.27
GENETICS
A genetic component of this disease has been recognized since at least28: 60% of patients have a somatic mutation and 40% have a genomic mutation.3 Of new patients, 6% to 10% have positive family histories, and 90% to 94% present as sporadic cases. A patient with bilateral retinoblastoma carries a heritable mutation in the germline. Preferential paternal transmission of the retinoblastoma gene (RB1) occurs in some kindreds.29 This is transmitted with a penetrance of 90%.3 Of unilateral cases, up to 10% to 15% of unilateral retinoblastoma cases may be hereditary and transmitted in an autosomal-dominant fashion.30,31
Two mutations are required to inactivate two alleles of a single locus.32 Knudson20 performed a retrospective statistical analysis and plotted logarithmically the proportion of cases not yet diagnosed versus age for bilateral and unilateral retinoblastoma. The result was a straight line for bilateral retinoblastoma, which indicated a single random somatic event for tumor induction. The unilateral cases demonstrated a curve of second-order kinetics, indicating that two somatic events were required. With a mean of three tumors per person, Poisson distribution showed that the tumorinitiating events were random and independent. The chance of a somatic event occurring in at least one cell in one person is approximately 95%. The chance of a somatic mutation occurring in a particular cell is approximately 5 × 10-6. The chance of two events occurring in the same cell is the square of the probability of one event, or 2.5 × 10-11. Heritable retinoblastoma has one mutation in the germline at birth. Therefore, the tumor is transmitted in a highly penetrant autosomal-dominant fashion.
The RB1 locus was identified through cytogenetic deletions of chromosome 13 and linkage to esterase D.33 Approximately 5% of retinoblastoma patients demonstrate a karyotypic deletion in chromosome 13q14.2 RB1 was first cloned in 1986 by Friend and associates.34 The RB1 gene spans 200 kilobases of DNA.35 Germline mutations in bilateral retinoblastoma patients with positive family histories without deletions established the RB1 cDNA as the retinoblastoma gene.36 RB1 gene alterations have been described in small cell lung carcinoma,37 B-cell chronic lymphocytic leukemia,38 breast carcinoma,39 bladder carcinoma,40 glioblastoma,41 and adrenocortical carcinoma.42 Other sporadic chromosomal abnormalities reported in patients with retinoblastoma occur on chromosomes 1, 6,43 11,44 and 13.
Chromosome evaluation has played a role in prenatal diagnosis23 and assessment of other family members at risk.4 Genetic counseling includes risk assessment. The risk of retinoblastoma in offspring (0.4), twins (0.9), and siblings (0.04) of index bilateral cases warrants frequent clinical examinations or examinations under anesthesia (EUA).45
RETINOBLASTOMA GENE PRODUCT
RB1 encodes a nuclear phosphoprotein p110RB1 with a molecular weight of 105 to 110 kilodaltons.46 In the G1 phase of the cell cycle, the protein is hypophosphorylated; in S, G2, and M, it is hyperphosphorylated. Known functions of p110RB1 include inhibition of cell cycle progression, inhibition of cell proliferation by TGF-beta, interference with DNA replication, and suppression of tumorigenicity.46 Although the exact mechanism is unknown, the protein acts as a nuclear regulator of cell proliferation47 and differentiation.46 It has been shown to interfere with RNA polymerase I transcription factor.48
CLINICAL PRESENTATION
Ocular presentation of retinoblastoma may be unilateral (70%) or bilateral (30%). In the United States, the age of onset in bilateral disease is 13 months and in unilateral disease 25 months.15 With a positive family history, mean age of onset for bilateral and unilateral presentations is less than 12 months. The mean age of diagnosis is advanced in many other countries.49,50,51
With asynchronous bilateral retinoblastoma, tumor in the second eye occurs an average of 11 months, but in some reports as late as 4 to 5 years, after diagnosis of tumor in the first eye.52,53 Leukocoria (50% to 60%) and strabismus (18% to 22%) are the most common presentations (Fig. 1).15 Only 3% of patients are diagnosed during a routine pediatric examination. Unusual presentations include orbital cellulitis,54 neovascular glaucoma,55 pseudohypopyon,56 and hyphema.57,58
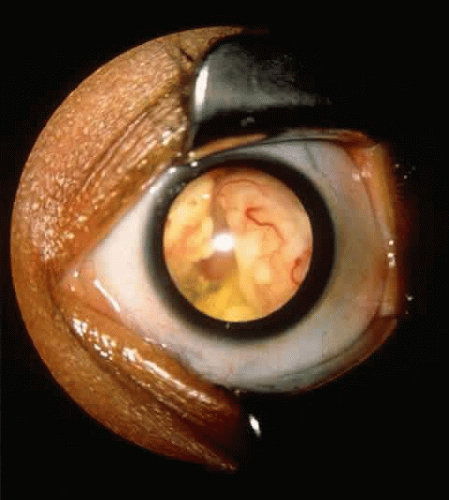 Fig. 1. Retinoblastoma presenting as leukocoria. |
Trilateral retinoblastoma is a rare variant of bilateral disease, with additional tumor in the pineal gland first described in59 Rare pineal involvement in unilateral cases has been reported.60 The pineal gland arises from the neuroectoderm and has diurnal neurosecretory properties.61 This is consistent with its role as a photoreceptor organ in lower vertebrates.62 The incidence of trilateral disease has been reported in the range of 3%60 to 6%,63 but it is probably less than 1%. The pineal tumor may be present at diagnosis of retinoblastoma,60 or it may occur up to 8 years later.64 The intracranial tumor may present first. Ectopic intracranial locations have been reported.65 Most trilateral patients have a positive family history of hereditary retinoblastoma.64
Several tumor growth patterns occur in all retinoblastomas. Endophytic growth (61%) occurs when the tumor grows into the eye relative to the retina. Ophthalmoscopy shows that this tumor sits on the retina (Fig. 2). Exophytic growth (39%) occurs when the tumor grows toward the choroid relative to the retina. Presentation is subretinal.66 Exophytic growth is associated with glaucoma and choroidal invasion. Diffuse infiltration of retinoblastoma represents up to 2% of retinoblastoma cases.67 Retinoma is a benign or spontaneously regressed variant representing less than 2% of all retinoblastomas.68 Malignant transformation has been reported.69 Retinoma may represent a heritable mutation.70
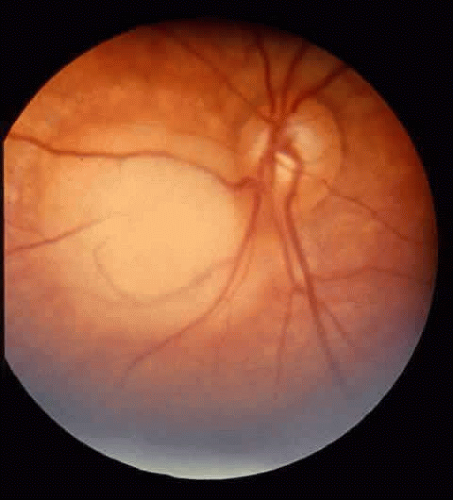 Fig. 2. Localized retinal lesion. |
Deletion of the long arm of chromosome 13 (the 13 q-deletion syndrome) may have systemic implications. The syndrome consists of microcephaly, broad nasal bridge, hypertelorism, microophthalmia, epicanthal folds, ptosis, micrognathia, short neck, low-set ears, anal abnormalities, and hypoplastic thumbs.71 Developmental delay and failure to thrive may occur. In these patients, evaluation of other chromosomal or infectious etiologies is important.72
DIFFERENTIAL DIAGNOSIS
Ocular abnormalities associated with leukocoria15 include retinoblastoma, persistent hyperplastic primary vitreous, Coats’ disease, retinopathy of prematurity, cataracts, toxocariasis, chronic retinal detachment, retinal astrocytoma, previous vitreous hemorrhage,73 and endogenous endophthalmitis with calcification.74 Imaging studies to aid in the diagnosis include computed tomography (CT), magnetic resonance imaging (MRI), and B-scan ultrasonography.73,75,76 CT effectively identifies both intraocular calcification and intracranial involvement (Fig. 3). MRI may be more effective in distinguishing Coats’ disease, persistent hyperplastic primary vitreous, and toxocariasis from retinoblastoma.73 Calcification may be seen with Coats’ disease77 or in rare cases may be absent with retinoblastoma.78
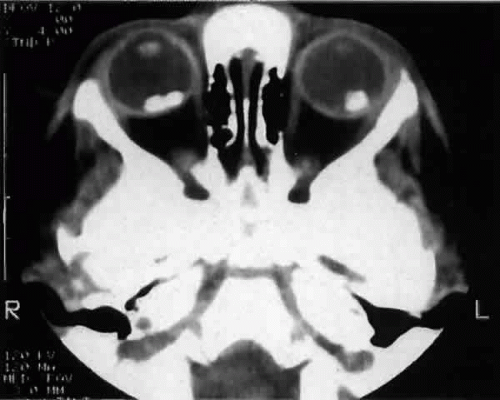 Fig. 3. Computed tomographic scan of a bilateral calcified retinoblastoma. |
Infrequently used diagnostic methods include aqueous lactate dehydrogenase activity and aspiration biopsy. The ratio of aqueous humor to serum lactate dehydrogenase is normal in at least 7% of retinoblastoma eyes.79 Aspiration cytology is a reliable tool for diagnosing extraocular retinoblastoma.80,81 Intraocular biopsy, however, carries a significant risk of extraocular spread along the needle tract.82 Other modalities reported include aqueous neuron-specific enolase,83 I123 metaiodobenzylguanidine scintigraphy,84 and proton magnetic resonance spectroscopy.85
CLASSIFICATION
Several tumor classification systems have been developed. The most commonly quoted one was proposed by Reese and Ellsworth (Table 1).15 The classification predicts the chance of globe salvage when treated with lateral port external beam radiation. The staging system considers tumor location and size. Other treatment modalities or levels of visual function are not considered. The Essen classification86 accounts for macular function and visual outcome. The St. Jude staging system considers extraocular extent and may be useful for determining prognosis. It considers retinal, extraretinal, extrachoroidal, and distant disease.87
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


