Retinal Degenerations and Dystrophies
Mithlesh C. Sharma
Allen C. Ho
Best’s Disease
Best’s disease, also known as vitelliform macular dystrophy, is a macular dystrophy that is clinically characterized by an egg yolk-like lesion at the level of the retinal pigment epithelium (RPE), usually located in the posterior pole. The disease progresses through various stages to culminate in macular atrophy or scarring with loss of central vision.
Epidemiology and Etiology
Symptoms develop in infancy or early childhood.
An excessive amount of lipofuscin-like material within the retinal pigment epithelial cells, particularly in the fovea, is observed. There appears to be a secondary loss of the photoreceptor cells.
It is an autosomal-dominant disease whose gene is mapped to chromosome 11q13.
Clinical Signs
Five stages are delineated.
Previtelliform stage: Patients are asymptomatic with no fundus abnormality, but electrooculography shows a reduced light-peak to dark-trough ratio.
Vitelliform stage: This stage is characterized by an egg yolk-like lesion in the macular area. It is usually detected during the first or second decade of life. Although usually single and bilaterally symmetric, multiple egg yolk lesions may be observed in the posterior pole. The egg yolk lesions are located at the level of the RPE, are rounded or oval in shape with distinct borders, and range from one-half to two disc diameters in size. Vision may be normal or slightly decreased at this stage (Fig. 5-1).
Pseudohypopyon stage: By the second or third decade of life, lesions break through the RPE, and the yellow material accumulates inferiorly in the macula
within the subretinal space to form pseudohypopyon (Fig. 5-2).
Vitelliruptive stage: The egg yolk breaks up to produce a scrambled egg appearance. Patients usually notice some visual impairment at this stage (Fig. 5-3).
End stage: Subretinal fibrosis, or a vascularized scar with choroidal neovascularization, contributes to the visual loss at this stage.
The vitelliform degeneration presenting after childhood is called adult Best’s disease. In the latter variant, the yellow foveal deposits are symmetric and similar to childhood Best’s disease except that the lesions are smaller and have a central pigmented spot. The most common lesion mistaken for Best’s disease is a yellow premacular hemorrhage (Fig. 5-4).
Diagnostic Evaluation
Visual acuity: Vision remains good while the egg yolk lesions are intact. Disruption or scarring of the lesions may reduce visual acuity to the level of 20/200.
Visual fields: Central visual fields are normal initially, but a relative scotoma may develop with time.
Color vision: Mild dyschromatopsia may be noticed.
Dark adaptometry is normal.
Electroretinography is normal.
Electrooculography: Best’s disease is one of the few conditions that results in an abnormal electrooculogram (EOG) in the setting of a normal electroretinogram (ERG). During an EOG, the light-peak:dark-trough ratio (Arden ratio) is typically below 1.5.
Fluorescein angiography: In the vitelliform stage, complete blockage of background choroidal fluorescence by the lesion is observed. Areas of hyperfluorescence due to atrophic RPE are noticed as the egg yolk lesions show disruption.
Prognosis and Management
In general, the overall prognosis is good as most patients retain reading level of vision in at least one eye throughout life.
When severe vision loss does take place in an eye, it occurs slowly and usually begins after the age of 40 years.
No treatment is available for Best’s disease.
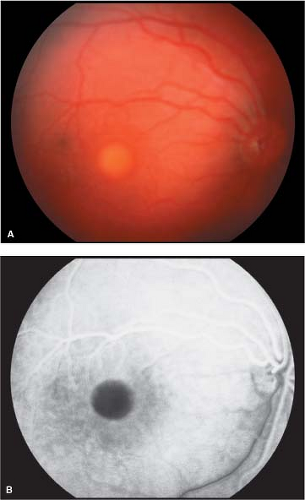 Figure 5-1. Best’s disease, vitelliform stage. A. Characteristic egg yolk-like lesion in the fovea. B. Corresponding fluorescein angiogram showing blocked fluorescence due to egg yolk lesion during transit phase. (Courtesy of Retina Slide Collection, Wills Eye Hospital, Philadelphia, Pennsylvania, compiled by Dr. Tamara Vrabec and Dr. Gordon Byrnes.) |
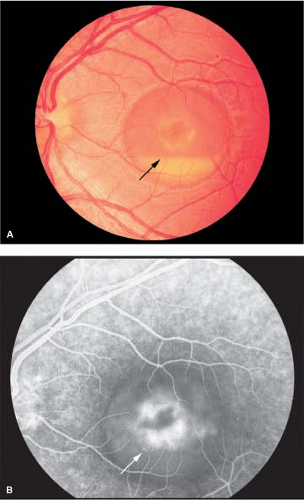 Figure 5-2. Best’s disease, pseudohypopyon stage. A. Collection of yellow material within the subretinal space simulating hypopyon (arrow). B. Blocked fluorescence (arrow) due to deposition of yellow material inferiorly and perifoveal hyperfluorescence in the center of the lesion is seen in fluorescein angiogram photographs. Courtesy of Retina Slide Collection, Wills Eye Hospital, Philadelphia, Pennsylvania, compiled by Dr. Tamara Vrabec and Dr. Gordon Byrnes.) |
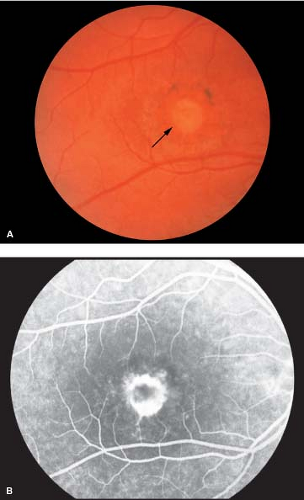 Figure 5-3. Best’s disease, vitelliruptive stage. A. Irregular areas of retinal pigment epithelial loss secondary to breakup of the egg yolk lesion (arrow). B. Intense perifoveal hyperfluorescence surrounded by multiple areas of hyperfluorescence is shown in the corresponding fluorescein angiogram. (Courtesy of Retina Slide Collection, Wills Eye Hospital, Philadelphia, Pennsylvania, compiled by Dr. Tamara Vrabec and Dr. Gordon Byrnes.) |
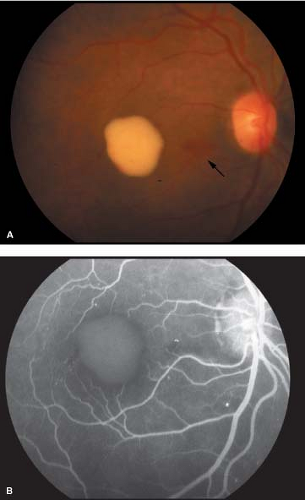 Figure 5-4. Pseudo–Best’s disease. A. Old premacular hemorrhage simulating the egg yolk lesion of Best’s disease. Note that the retinal vessels are obscured by the preretinal lesion (arrow). Slightly irregular borders of the lesion and presence of neighboring retinal hemorrhages are clues to the yellow lesion being an old hemorrhage. B. Fluorescein angiogram demonstrating microaneurysms and retinal telangiectasia temporal to the lesion, consistent with diabetic retinopathy and a premacular hemorrhage. |
Cone Dystrophy
Cone dystrophy is an inherited defect that primarily affects the cone photoreceptor system.
Epidemiology and Etiology
Symptoms begin in early childhood to middle adulthood.
Cone dystrophy is primarily autosomal dominant, although autosomal-recessive and X-linked forms have also been reported.
History
The rate of progression and the severity of signs and symptoms are variable. The symptoms consist of progressive visual loss, hemeralopia (decreased vision in brightly illuminated environment), color vision difficulties, and central visual field defects.
Macular changes typically follow the visual disturbances; therefore, early in the disease process the fundus may appear entirely normal.
Clinical Signs
There is gradual, typically symmetric loss of visual acuity to the level of 20/200. Occasionally, this may be reduced to the level of counting fingers to hand motion acuity.
Fundus changes are variable (Fig. 5-5). Early in the disease process, pigmentary stippling with diffuse pigment granularity in the posterior pole is the most common abnormality observed. The classic “bull’s-eye” pattern of retinal pigment epithelial atrophy is a late finding. In advanced cases, a round, discrete area of central atrophy is seen. Temporal pallor of the optic disc may be observed.
Differential Diagnosis
Reduced vision with normal fundus in children: In this category, cone dystrophy should be differentiated from Stargardt’s disease.
“Bull’s-eye” maculopathy: The following causes of “bull’s-eye” maculopathy need to be considered in the differential diagnosis:
Stargardt’s disease
Chloroquine toxicity
Batten’s disease
Benign concentric annular macular dystrophy
Leber’s congenital amaurosis
Diagnostic Evaluation
Visual fields: Central scotoma is usually seen.
Color vision: Reduced.
Dark adaptometry: The cone component of the dark adaptation curve is abnormal.
Electroretinography: The single-flash photopic ERG and the photopic flicker ERG are low or unrecordable. The scotopic ERG is usually normal.
Fluorescein angiography: The retinal pigment epithelial changes in the macular area may be visible by fluorescein angiography before they can be visualized clinically. Early in the disease process, a mottled hyperfluorescence is seen. As the “bull’s-eye” pattern of retinal pigment epithelial loss develops, hyperfluorescence surrounding a central area of hypofluorescence is observed.
Prognosis and Management
The visual loss is gradual and symmetric to the level of 20/200. However, it may occasionally be severe enough to cause a visual acuity of counting fingers to hand motion. The visual loss is more severe in early-onset cases.
No treatment is available for cone dystrophy.
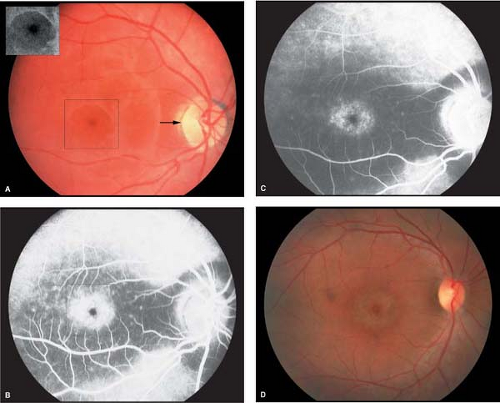 Figure 5-5. Cone dystrophy. A. Early “bull’s-eye” maculopathy in a patient with cone dystrophy (inset). Note the temporal optic disc atrophy (arrow). B. Corresponding fluorescein angiogram showing central hypofluorescence surrounded by a ring of hyperfluorescence seen during the transit phase (box). Cone dystrophy. C. The hyperfluorescence fades away in the later frame indicating the presence of window defects due to retinal pigment epithelial atrophy. D. Advanced cone dystrophy with a classic “bull’s-eye” maculopathy. Note the temporal optic disc pallor. (Courtesy of Dr. Joseph Maguire and the Retina Slide Collection, Wills Eye Hospital, Philadelphia, Pennsylvania, compiled by Dr. Tamara Vrabec and Dr. Gordon Byrnes.) |
Pattern Dystrophy
Pattern dystrophy describes a group of related conditions that are inherited in an autosomal-dominant fashion and are clinically characterized by variably shaped yellow or gray deposits in the macula.
Epidemiology and Etiology
Symptoms begin in middle age.
The disease has an autosomal-dominant pattern. Genetic analysis of patients with butterfly dystrophy has shown mutation in the peripherin/RDS gene, located on the short arm of chromosome 6. The peripherin gene product plays an important role in the structural integrity of photoreceptor outer segment discs. However, this mutation does not correspond to a particular phenotype. That is, other forms of retinal degeneration have been linked to peripherin/RDS gene mutations.
History
Most patients are either asymptomatic or have minimal visual disturbances. Typically, the diagnosis is made on routine fundus examination of a middle-aged adult.
Clinical Signs
Visual acuity: Patients may have normal visual acuity up to the fifth or sixth decade of life. Reduced vision and metamorphopsia may be the presenting symptoms.
Ophthalmoscopically: The following patterns of pigment deposits in the macular area may be observed:
Most commonly, a bilateral, triradiate (“butterfly”) pattern of yellow or gray pigment at the level of the RPE in the central macular region. A rim of retinal pigment epithelial atrophy around the pigment figure, which is more apparent on fluorescein angiography, may also be seen (Figs. 5-6 and 5-7).
A single, round, vitelliform lesion in the fovea (adult-onset foveomacular vitelliform dystrophy).
Extensive macular fishnet arrangement (reticular dystrophy).
Coarse pigment mottling of the macula (fundus pulverulentus) (Fig. 5-8).
The affected members of a given pedigree may have different patterns, and the patterns may differ in the eyes of an affected individual. Even a change from one to another pattern over time may be observed.
Differential Diagnosis
Large drusen: Ophthalmoscopically, the yellow pigment figures of pattern dystrophy may be confused with the large drusen of age-related macular degeneration.
Diagnostic Evaluation
Visual fields: Normal, except for minimally reduced sensitivity in the macular area.
Color vision, dark adaptometry, electroretinography: Normal.
Electrooculography: Mildly abnormal, which is consistent with the disturbed retinal pigment epithelial function.
Fluorescein angiography: Pigment figures are hypofluorescent throughout the study. The retinal pigment epithelial atrophy around the lesions produces hyperfluorescence.
Prognosis
The prognosis for retention of good central vision in at least one eye throughout life is excellent.
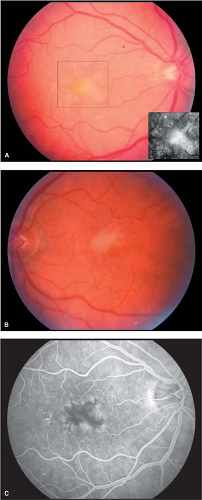 Figure 5-6. Pattern dystrophy. A and B. Bilateral, multiple, discrete, yellow areas at the level of the retinal pigment epithelium (RPE) in the central macular region (inset). Pattern dystrophy. C. The lesions are more pronounced in the corresponding fluorescein angiogram image of the right eye, where there is patchy hyperfluorescence. |
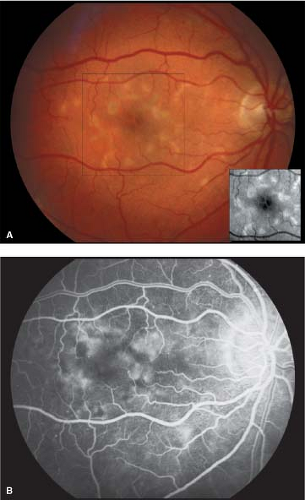 Figure 5-7. Pattern dystrophy. A. Classic triradiate (“butterfly”) pattern of pigment deposits at the fovea (inset) surrounded by multiple areas of retinal pigment epithelial atrophy. B. Fluorescein angiogram showing patchy hyperfluorescence in the arteriovenous phase. The hyperfluorescent areas correspond to retinal pigment epithelial atrophy in the macular region. (Courtesy of Dr. Eric Shakin and the Retina Slide Collection, Wills Eye Hospital, Philadelphia, Pennsylvania, compiled by Dr. Tamara Vrabec and Dr. Gordon Byrnes.) |
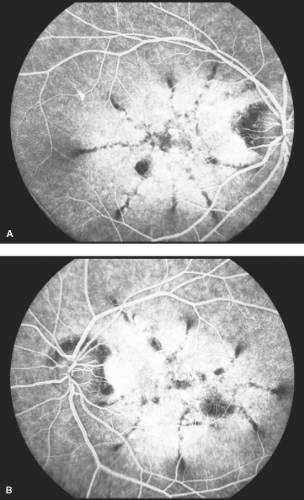 Figure 5-8. Pattern dystrophy, fundus pulverulentus. Fluorescein angiogram photographs of both eyes showing radiating pattern of hypofluorescence due to coarse pigment deposits at the level of RPE—reticular dystrophy. (Courtesy of Dr. William Annesley, and the Retina Slide Collection, Wills Eye Hospital, Philadelphia, Pennsylvania, compiled by Dr. Tamara Vrabec and Dr. Gordon Byrnes.) |
Stargardt’s Disease
Stargardt’s disease is a macular dystrophy that is characterized by the presence of discrete, yellow, pisciform flecks at the level of the RPE. Currently, Stargardt’s disease and fundus flavimaculatus are regarded as variants of the same disorder. The term fundus flavimaculatus is generally applied when the characteristic flecks are scattered throughout the fundus. When the flecks are confined to the posterior pole and are associated with macular atrophy, the condition is described as Stargardt’s disease.
Epidemiology and Etiology
The disease usually presents in the first or second decade of life.
Both sexes are affected equally.
Stargardt’s disease is usually autosomal recessive, although dominantly inherited cases have been described. The gene for autosomal-recessive Stargardt’s disease is located on chromosome 1. This gene codes for an ATP-binding transport protein (ABCR) that is expressed in the rod inner segments, but not the RPE. A homozygous mutation in the ABCR gene causes fundus flavimaculatus.
History
Children with Stargardt’s disease are usually brought to the attention of an ophthalmologist as a result of a gradual impairment of vision noticed by the parents or after failing a school vision screening.
Clinical Signs
Visual acuity is slightly affected in the beginning but may be severely reduced in the later stage of disease. Loss of the foveal reflex may be the only initial clinical finding. Discrete, yellowish, “pisciform” flecks located at the level of the RPE are often noticed at some point in the course of disease. The flecks may or may not involve the macula (Fig. 5-9). Perifoveal retinal pigment epithelial mottling may become evident with the progression of disease.
A “bull’s-eye” pattern of retinal pigment epithelial loss may become apparent, particularly by fluorescein angiography. The macula classically develops a “beaten bronze” appearance corresponding to atrophy of the central RPE in the advanced stage of disease (Figs. 5-10 and 5-11). Histopathology shows an accumulation of an abnormal lipofuscin-like material in the RPE (Fig. 5-12).
Differential Diagnosis
Cone dystrophy: reduced vision and normal fundus in a child
“Bull’s-eye” maculopathy: chloroquine toxicity, Batten’s disease, benign concentric annular macular dystrophy
Diagnostic Evaluation
Visual fields: Usually a central scotoma is noted, but a paracentral scotoma, central constriction, and a ring scotoma may also be seen, especially early in the disease.
Color vision: Mild dyschromatopsia to red and green may be noted.
Dark adaptometry: Dark adaptation may be delayed.
Fluorescein angiography: Features that may help confirm the diagnosis of Stargardt’s disease include dark or silent choroid;
multiple, irregular hyperfluorescent spots that do not precisely correspond to the flecks; and a “bull’s-eye” window-defect pattern of hyperfluorescence in the macula.
Electroretinography: Usually normal but may be reduced with increasing amounts of peripheral flecks and atrophy.
Electrooculography: Usually subnormal.
Prognosis and Management
The majority of patients preserve moderate visual acuity (20/70 to 20/200), at least in one eye.
No effective treatment is available for Stargardt’s disease.
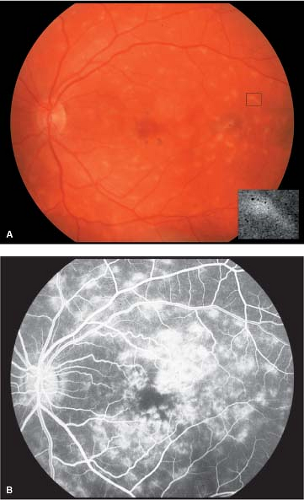 Figure 5-9. Stargardt’s disease. A. Multiple, discrete, yellow, “pisciform” flecks (inset shows one fleck) located at the level of the RPE with corresponding hyperfluorescent areas in the fluorescein angiogram photograph are distributed throughout the posterior pole of the left eye. B. The background choroid is dark on the fluorescein angiogram photograph, and there is transmission hyperfluorescence associated with macular flecks and retinal pigment epithelial alterations. (Courtesy of Retina Slide Collection, Wills Eye Hospital, Philadelphia, Pennsylvania, compiled by Dr. Tamara Vrabec and Dr. Gordon Byrnes.) |
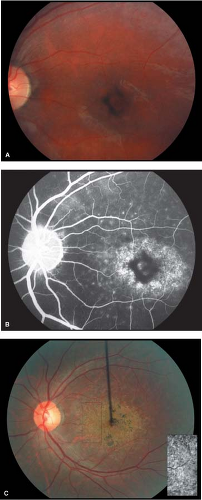 Figure 5-10. Stargardt’s disease. A. Advanced Stargardt’s disease with “beaten bronze” macula. B. Corresponding fluorescein angiogram showing a central area of hypofluorescence (retinal pigment epithelial clumping) surrounded by a ring of hyperfluorescence (retinal pigment epithelial atrophy). Note the dark or silent choroid (blocked fluorescence). Stargardt’s disease. C. Stargardt’s disease with “bull’s-eye” macula. Compare with Figure 5-5D. Note the “beaten bronze” appearance of the macula (inset). (Courtesy of Dr. Eric Shakin and the Retina Slide Collection, Wills Eye Hospital, Philadelphia, Pennsylvania, compiled by Dr. Tamara Vrabec and Dr. Gordon Byrnes.)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|