Retinal and Choroidal Tumors
Franco M. Recchia
Astrocytic Hamartoma
Astrocytic hamartoma is a congenital, minimally progressive, benign tumor arising from the glial cells of the retina and usually located around the optic disc. It is often associated with the systemic condition tuberous sclerosis (Bourneville’s disease), occasionally with neurofibromatosis, but also occurs sporadically in otherwise normal individuals.
Epidemiology and Etiology
Most astrocytic hamartomas occur congenitally in association with tuberous sclerosis (TS), a familial phakomatosis characterized by the triad of seizures, mental retardation, and skin lesions. TS has an estimated incidence of 1 in 15,000 to 1 in 100,000 and exhibits autosomal-dominant inheritance. Roughly half of the patients with TS have astrocytic hamartomas. Causative genes have been identified on chromosomes 9q34 and 16p13.
History
Patients with astrocytic hamartomas are usually asymptomatic. Visual field testing may reveal a scotoma in the area corresponding to the tumor.
Important Clinical Signs
The appearance of a retinal astrocytic hamartoma is principally of two types:
Smaller, noncalcified, flat, smooth tumor that appears as mild thickening of the nerve fiber layer
Larger, calcified, whitish-yellow nodular mass (“mulberry lesion”)
Aspects of both may be seen in the same lesion, as it is likely that calcification progresses slowly over many years (Fig. 6-1).
Every patient with an astrocytic hamartoma of the retina or optic nerve must be evaluated for TS. Typical manifestations of TS include:
Skin lesions (∼95% incidence)
Hypomelanotic macules: Oval (“ash-leaf”), polygonal, or punctate (“confetti”) in shape. Usually present at birth and often the first presenting sign.
Reddish-brown papular rash over the face (termed adenoma sebaceum, but actually angiofibromas), often mistaken for acne. Rarely present before a few years of age.
Seizures (∼90% incidence): Myoclonic spasms lasting 10 to 50 seconds; tend to become grand mal type later in life.
Mental retardation (∼60% incidence)
Associated Clinical Signs
Ocular: Occasionally, may produce vitreous hemorrhage, vitreous seeding, subretinal hemorrhage, or retinal detachment.
Systemic (additional manifestations of TS): Ungual fibromas, pleural cysts (leading to spontaneous pneumothorax), renal angiomyolipoma, cardiac rhabdomyoma, and hamartomas of the liver, thyroid, pancreas, or testis may occur.
Differential Diagnosis
Retinoblastoma (Rb)
Myelinated nerve fibers
Retinal granuloma
Drusen of the optic disc
Papillitis
Diagnostic Evaluation
Fluorescein angiography: The tumor appears relatively hypofluorescent in the arterial phase. Superficial fine blood vessels are seen during the venous phase. The tumor stains intensely and homogeneously in the late phases.
B-scan Ultrasonography: A larger, calcified lesion often appears as a discrete, oval, solid mass with a sharp anterior border.
Neuroimaging: Subependymal hamartomas, characteristic of TS, may be seen with computed tomography (CT) or magnetic resonance imaging (MRI).
Prognosis and Management
The vast majority of retinal astrocytic hamartomas are asymptomatic and do not require treatment.
Associated retinal detachment can often be treated with demarcating laser photocoagulation.
Patients and family members should be examined regularly for manifestations of TS.
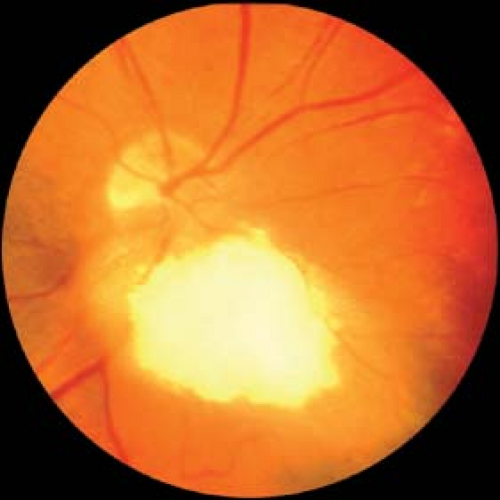 Figure 6-1. Astrocytic hamartoma. A peripapillary astrocytic hamartoma can be seen with classic mulberry calcification overlying an underlying smooth tumor. Patients and family members should be examined for tuberous sclerosis. Courtesy of Drs. Jerry and Carol Shields, Oncology Service, Wills Eye Hospital, Philadelphia, Pennsylvania. |
Retinoblastoma
Rb is the most common intraocular malignancy of children. It may be familial but most often is sporadic in occurrence. The tumor arises from cells in the developing retina of one or both eyes as a result of mutations in the Rb) tumor-suppressor gene. Rb presents most often as a white intraocular mass with propensity for direct extension into the brain and almost certain mortality if untreated.
Epidemiology and Etiology
Rb occurs in approximately 1 in 15,000 live births. Most children are diagnosed at a mean of 18 months.
No predilection for race or gender has been shown.
Roughly two-thirds of the cases are unilateral, and one-third is bilateral. Unilateral cases are more likely to be diagnosed at an older age (mean of 24 months) and are most often nonfamilial (sporadic). Bilateral cases are diagnosed at a mean of 12 months, are usually familial, and are nearly always multifocal.
Rb results from mutations or loss of both alleles of the Rb gene, located on chromosome 13q14. The Rb gene product appears to function as regulator of cellular proliferation through inhibitory effects on gene transcription at specific stages of the cell cycle. The timing of allelic inactivation determines whether the mutation is germinal (i.e., heritable by offspring of the affected child) or somatic (nonheritable). In germinal cases, a mutant allele is present before fertilization, most commonly as a result of inheritance from either parent. In somatic cases, both alleles are present and active at fertilization, and spontaneous mutations in each allele arise subsequently.
History
A white pupillary reflex (leukokoria; Fig. 6-2) and strabismus are the two most common findings reported by parents. In fewer than 10% of cases is a family history known at the time of diagnosis.
Important Clinical Signs
Most patients present with leukokoria and strabismus. Small retinoblastomas appear as flat, translucent, white retinal lesions (Fig. 6-3A). With growth, the tumor appears more solid, elevated, and chalky-white, with overlying dilated tortuous blood vessels.
Three growth patterns have been described.
Endophytic: The tumor grows from the retina inward to seed the vitreous cavity or anterior chamber.
Exophytic: The tumor grows from the retina outward to occupy the subretinal space, often causing an exudative retinal detachment (Fig. 6-3B).
Diffuse infiltrating: The least common form; this is characterized by shallow spread of tumor along the entire retina and into the vitreous and anterior chamber.
Other important findings are iris neovascularization, which occurs in nearly one-fifth of all cases, and “pseudohypopyon” (settling of tumor and inflammatory cells in the anterior chamber).
Associated Clinical Signs
Differential Diagnosis
Leukokoria
Coats’ disease
Persistent fetal vasculature syndrome (formerly termed persistent hyperplastic primary vitreous, or PHPV)
Toxocariasis
Retinopathy of prematurity
Familial exudative vitreoretinopathy
Retinal astrocytoma
Cataract
Norrie’s disease
Incontinentia pigmenti
Vitreous seeding
Intraocular inflammation
Endophthalmitis
Vitreous hemorrhage
Leukemic infiltration
Diagnostic Evaluation
Detailed systemic evaluation and examination is required, as well as family history and ocular examination of parents, and complete examination of both eyes (often requiring anesthesia for complete visualization of the fundi with scleral depression).
Ultrasonography: An elevated, rounded, intraocular mass is seen, with high internal reflectivity (calcification) and shadowing of sclera and soft tissue posterior to the lesion.
CT is helpful in detecting intraocular calcification (present in roughly 80% of retinoblastomas), assessing extraocular extension, and identifying the presence of pineal tumors.
Fluorescein angiography reveals early arterial filling of the vessel feeding the tumor, leakage of dye from intrinsic tumor vessels, and late hyperfluorescence of the tumor.
Prognosis
Spontaneous regression is rare and leads to phthisis bulbi. Typically, if untreated, children die within 2 years of diagnosis. Early detection, coupled with improvements and promptness of treatment, has reduced the mortality rate to less than 10%. The main determinant for mortality is optic nerve invasion. For this reason, it is imperative to obtain as long a section of optic nerve as possible during enucleation.
Prognostic factors for failure to preserve vision or to preserve the eye are larger tumor size, vitreous seeding, and macular involvement.
Children with germinal Rb have an increased risk of developing other primary malignancies over the course of their lifetimes. These tumors include principally intracranial Rb, osteogenic sarcoma of the long bones, and sarcoma of soft tissues. The risk is estimated to be 20% within 25 years of treatment.
Management
Children diagnosed with Rb should undergo evaluation for systemic involvement, including complete blood count, lumbar puncture, neuroimaging, and bone marrow biopsy. Genetic testing of the child and family members should be performed.
Individual treatment varies according to number, size, and location of tumors, as
well as systemic status. Therapeutic options include cryotherapy, laser photocoagulation, enucleation, external-beam irradiation, plaque radiotherapy, chemotherapy, thermotherapy, and chemothermotherapy.
Chemoreduction prior to definitive ocular treatment may be helpful in reducing the need for enucleation and reducing the rate of occurrence of pinealoblastoma.
In familial cases, genetic counseling provides parents with important information regarding the probability of further occurrences. Patients with germinal Rb must be warned of the possibility of transmission to their offspring.
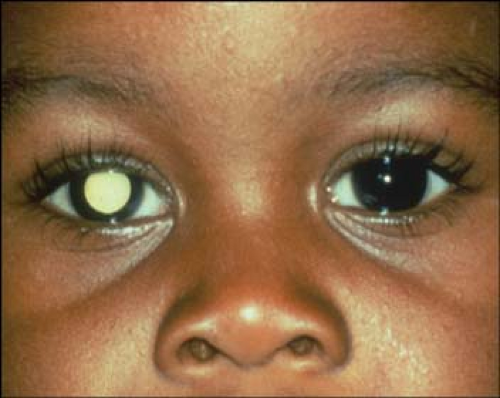 Figure 6-2. Retinoblastoma. This young boy has leukokoria and strabismus, the most common clinical presentation of retinoblastoma. Courtesy of Drs. Jerry and Carol Shields, Oncology Service, Wills Eye Hospital, Philadelphia, Pennsylvania. |
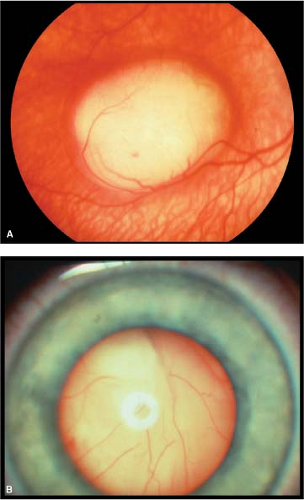 Figure 6-3. Retinoblastoma. A. Focal retinoblastoma presenting as a macular intraretinal amelanotic tumor. B. Massive exophytic retinoblastoma with tumor behind the clear lens. Courtesy of Drs. Jerry and Carol Shields, Oncology Service, Wills Eye Hospital, Philadelphia, Pennsylvania. |
Retinal Capillary Hemangioma
Originally termed angiomatosis retinae, retinal capillary hemangioma is a benign vascular tumor of variable size located in the retina or adjacent to the optic disc. Usually diagnosed by the fourth decade, it may be the first manifestation of von Hippel–Lindau (VHL) disease, a familial cancer syndrome with which it is commonly associated.
Epidemiology and Etiology
This tumor may occur in a sporadic or hereditary fashion. Retinal capillary hemangioma occurs in up to 80% of patients with VHL syndrome and is often the first manifestation, diagnosed at a mean age of 25 years.
The VHL syndrome has an estimated prevalence of 1 in 40,000 and is possibly more common in whites. It exhibits dominant inheritance and has variable phenotypes within families.
Mean survival of patients with VHL is 41 years of age.
VHL syndrome is caused by mutations in the VHL gene, a tumor-suppressor gene located on chromosome 3p25. The VHL gene product regulates the expression and function of hypoxia-responsive angiogenic factors [e.g., vascular endothelial growth factor (VEGF)].
History
Capillary hemangiomas may be diagnosed incidentally or in patients suspected of having VHL syndrome. The tumors may be asymptomatic or may produce painless visual impairment from vitreous hemorrhage, macular pucker, or retinal detachment.
Important Clinical Signs
Retinal capillary hemangioma: Usually located peripherally and well circumscribed. Initially appears as a yellow-red dot with a minimally dilated “feeding” arteriole or draining venule (Fig. 6-4). With growth, appears orange-red with more prominently dilated afferent and efferent vessels. May have associated exudation, subretinal fluid, or preretinal fibrosis.
Juxtapapillary capillary hemangioma: Orange-red in color, but less well circumscribed. It often lacks feeder vessels.
VHL syndrome: Hemangio-blastomas of the cerebellum and spinal cord, renal cell carcinoma, or pheochromocytoma.
Associated Clinical Signs
Retinal detachment and, rarely, neovascular glaucoma may complicate capillary hemangioma.
Hemangiomas of the adrenal glands, lungs, and liver, and multiple cysts of the pancreas and kidneys have been observed in some patients with VHL syndrome.
Differential Diagnosis
Retinal capillary hemangioma
Other tumors: Retinal cavernous hemangioma, racemose hemangioma, choroidal melanoma, and astrocytic hamartoma
Vascular diseases: Coats’ disease, retinal arterial macroaneurysm, familial exudative vitreoretinopathy, and exudative macular degeneration
N.B.: A distinction has been made between the retinal angioma of VHL
syndrome and an acquired, nonhereditary entity occurring in older patients and termed vasoproliferative retinal tumor. This latter condition is usually located in the inferotemporal peripheral fundus and lacks markedly dilated feeder vessels.
Optic disc hemangioma
Papillitis
Optic disc granuloma
Optic disc glioma
Diagnostic Evaluation
Ocular: Fluorescein angiography is the most helpful ancillary study. In the arterial phase, a prominent, dilated feeder arteriole may be seen. The tumor appears hyperfluorescent early and remains so through the late phases, sometimes leaking dye into the vitreous. Ultrasonography may be helpful in diagnosing lesions of greater than 1 mm and demonstrates acoustic solidity throughout the lesion.
Systemic: All patients with retinal capillary hemangioma, as well as relatives, should be evaluated for VHL syndrome. Genetic testing for mutations in the VHL gene is available.
Prognosis and Management
The natural history is variable, with both progressive enlargement and spontaneous regression having been reported. Visual prognosis is highly variable and depends on tumor location, size, and associated complications, especially those that involve the macula.
In patients with VHL syndrome, risk of developing associated tumors increases with age. Morbidity and mortality are related to these associated tumors. Median survival is less than 50 years, with renal carcinoma as the leading cause of death.
Treatment is recommended for tumors with documented growth or effects on visual function. The goal of treatment is to induce resolution of exudation or subretinal fluid. Laser photocoagulation is reserved for smaller tumors (less than 2 mm in diameter). Larger tumors are best treated with cryotherapy or plaque radiotherapy. Vitreoretinal surgery, and, rarely, enucleation may be necessary for patients with advanced or uncontrollable pathology.
Patients with VHL syndrome and their relatives should be examined regularly for life. This includes regular testing for urine catecholamines, CT and ultrasonography of the kidneys, and MRI of the brain.
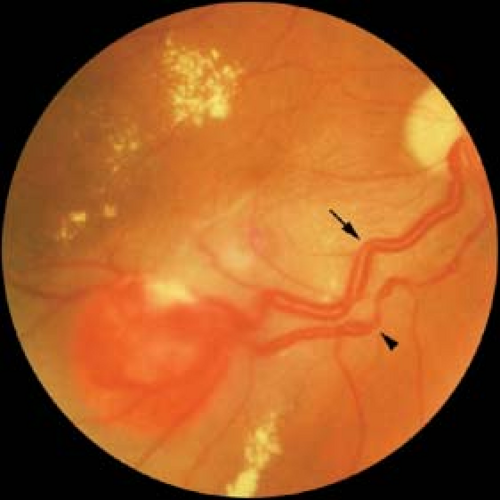 Figure 6-4. Retinal capillary hemangioma. Note the dilated feeding retinal arteriole (arrow) and the segmented draining retinal vein (arrowhead) with associated macular edema, premacular fibrosis, and lipid exudation. These lesions may be the first manifestation of von Hippel–Lindau disease. Courtesy of Drs. Jerry and Carol Shields, Oncology Service, Wills Eye Hospital, Philadelphia, Pennsylvania. |
Retinal Cavernous Hemangioma
Retinal cavernous hemangioma is a benign, rarely progressive, vascular tumor of the retina or optic disc characterized by a collection of venous aneurysms. It may be associated with similar vascular anomalies of the skin and central nervous system (CNS).
Epidemiology and Etiology
Tumor occurrence is mostly sporadic.
Small pedigrees of patients with cavernous hemangioma as part of a dominantly inherited oculoneurocutaneous syndrome have been reported.
History
Patients are usually asymptomatic, but the tumor may produce painless visual loss.
A hereditary component may be noted.
Important Clinical Signs
A cluster of dark-red, intraretinal aneurysms is located along a retinal venule, appearing as a “cluster of grapes” arising from the inner retinal surface (Fig. 6-5). Often there is overlying gray fibroglial tissue. Usually the tumor does not have associated exudation or a feeding arteriole.
Associated Clinical Signs
Vitreous hemorrhage occurs in up to 10% of cases.
There may also be cutaneous vascular malformations.
Cavernous hemangiomas of the midbrain or cerebellum may produce seizures or subarachnoid hemorrhage.
Differential Diagnosis
Capillary hemangioma
Acquired vasoproliferative tumor
Racemose hemangioma
Coats’ disease
Diagnostic Evaluation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


