9 Retina
An outpost of the central nervous system (CNS), the retina is a colony of brain cells that is located in the periphery of the body where it is able to interact with and detect the rather narrow spectrum of electromagnetic radiation called visible light. Death, destruction, or loss of all or part of the retina renders the eye sightless. Visual loss due to the death of retinal cells is permanent and irrevocable, because, like brain cells, the cells of the retina are incapable of repair or regeneration. Retinal anatomy is discussed in detail in Chapter 1.
DEVELOPMENTAL ANOMALIES
Dysplasia of the retina is a characteristic manifestation of trisomy 13 and also occurs in the Walker-Warburg syndrome. Congenital nonattachment of the retina is a rare anomaly caused by faulty invagination of the optic vesicle. Congenital detachment of the retina causes leukocoria in male infants with X-linked Norrie disease, who also are deaf and mentally retarded. The retina lining a choroidal coloboma may be absent, hypoplastic, or dysplastic.
Aplasia of the fovea occurs in aniridia and albinism. Albinos also have a lightly pigmented albinotic fundus. Variants of oculocutaneous albinism are caused by recessively inherited mutations in four genes. Patients with OCA1a, the classic form of the so-called tyrosinase-negative oculocutuaneous albinism, have white hair, pink eyes, and pink irides that transilluminate vividly. OAC1a is caused by mutations in the tyrosinase gene on chromosome 11 (11q14-q21) that cause complete absence of tyrosinase enzyme activity. Some patients with less deleterious tyrosinase mutations (OCA1b) initially were classified as tyrosinase-positive albinos because they had some pigmentation. Temperature-sensitive tyrosinase mutations analogous to those responsible for the pigmented ears and limbs of Siamese cats cause peripheral pigmentation in some human albinos. Oculocutaneous albinism type 2 (OCA2) is another variant of tyrosinase-positive albinism that is very common in some African populations such as the Ibo of Nigeria, in whom the prevalence is 1/1,100. Affected patients have yellow hair and hazel irides. OCA2 is caused by mutations in the OCA2 or P gene on chromosome 15 (15q11.2-q12). OCA3 and OCA4 are caused by mutations in the genes for tyrosinase-related protein-1 (TYRP1) 9p23 and membrane-associated transporter protein (MATP), respectively. The Hermansky-Pudlak syndrome is common in Puerto Ricans who also have a bleeding diathesis and bruise easily. Parents of affected children have been wrongfully accused of child abuse. Albinos with the rare autosomal recessively inherited Chédiak-Higashi syndrome are subject to bacterial and fungal infection and a peculiar form of lymphoproliferation. They have abnormal leukocytes with giant lysosomes and macromelanosomes. The syndrome is caused by mutations in the lysosomal trafficking regulator gene (LYST) on chromosome 1 (1q42.1-q42.2).
Albinism may affect both the skin and eyes (oculocutaneous albinism) or the eyes primarily (ocular albinism). Ocular albinos typically present with decreased vision and nystagmus caused by foveal aplasia. Iris transillumination serves to identify ocular albinos, who have normal skin pigmentation and may have dark hair. Macromelanosomes are found in the retinal pigment epithelium (RPE) and skin in X-linked ocular albinism (Fig. 9-1). Skin biopsy occasionally is used to confirm the diagnosis. Although white female carriers of X-linked ocular albinism have normal vision, iris transillumination is present and ophthalmoscopy may disclose a “mud-spattered” appearance of the fundus caused by patchy depigmentation of the peripheral RPE. Most common, the X-linked or Nettleship-Falls type of ocular albinism is caused by mutations in the G-protein-coupled receptor 143 gene (GPR143) on the X chromosome (Xp22.3).
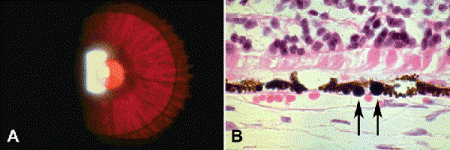
Fig. 9-1. Albinism. A. Transillumination of the iris discloses periphery of the lens and ciliary processes. B. X-linked ocular albinism. The RPE in postmortem eye contains large spherical macromelanosomes (arrows). (B. H&E ×250 [Microslide courtesy of Dr. W.R. Green, From Eagle RC Jr. Congenital, developmental and degenerative disorders of the iris and ciliary body. In: Albert DM, Jakobiec FA, eds. Principles and Practice of Ophthalmology. Clinical Practice, vol. 1. Philadelphia, PA: Saunders, 1993:367–389.])
Congenital hypertrophy of the RPE (CHRPE) (Fig. 11-24A,B) and congenital grouped pigmentation of the RPE are characterized by increased pigmentation and hypertrophy of RPE cells (Fig. 11-24C,D). See Chapter 11 below.
Myelinated nerve fibers occur in about 1% of the population. Myelination of optic nerve axons normally terminates at the posterior margin of the lamina cribrosa. The patches of aberrant myelination of the nerve fiber layer (NFL) may or may not be contiguous with the optic disc. Myelinated nerve fibers produce a focal scotoma.
Congenital vascular abnormalities of the retina include retinal arteriovenous communication, cavernous hemangioma of the retina, Leber miliary retinal aneurysms, Coats disease (Fig. 12-18), and parafoveal telangiectsias. Retinal hemangioblastomas may occur sporadically or in association with Von Hippel–Lindau disease (Figs. 2-10 and 2-11).
A peripheral retinal fold called Lange fold occurs at the ora serrata in infant eyes. Lange fold does not occur in vivo; it is a fixation artifact caused by vitreous traction.
RETINAL HEMORRHAGES AND EXUDATES
The retinopathies that occur in a variety of systemic and ocular diseases actually are composed of six types of retinal hemorrhages and two types of retinal exudates that occur in varying constellations.
The clinical appearance of retinal hemorrhages is determined by the location of blood in the retina (Fig. 9-2). Retinal hemorrhages are categorized as superficial (flame or splinter shaped), deep (blot and dot), sub-RPE, sub-internal limiting membrane (ILM), subhyaloid, and vitreous.
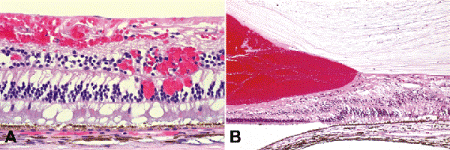
Fig. 9-2. Retinal hemorrhages. A. Flame or splinter hemorrhages are located in the inner nerve fiber layer of the retina. Blot and dot hemorrhages are located in the deeper retinal layers. B. Subhyaloid hemorrhage, proliferative diabetic retinopathy. Blood is located between the ILM and the posterior surface of the detached vitreous. (A. H&E ×100, B. H&E ×25)
Splinter- or flame-shaped hemorrhages are located superficially in the inner NFL of the retina (Fig. 9-2A). The configuration and feathery margin of these hemorrhages are caused by tracking of the erythrocytes along the axons of the ganglion cells as they arch above and below the fovea. The pattern of hemorrhages can graphically highlight the arcuate distribution of the nerve fibers in patients who have numerous superficial hemorrhages, for instance, after a branch retinal vein occlusion.
Blot and dot hemorrhages occur in the deeper retinal layers where the axons are oriented perpendicular to the plane of Bruch membrane. Here, the extravasations of blood have a discrete localized configuration because the erythrocytes are corralled or fenced-in by the surrounding axons.
Scaphoid or boat-shaped hemorrhages are preretinal hemorrhages that have a fluid level caused by settling-out of the red cells. Two types of scaphoid hemorrhages are recognized histopathologically: sub-ILM and subhyaloid hemorrhages. True subhyaloid hemorrhages are common in patients with proliferative diabetic retinopathy, in whom blood collects between the ILM and the posterior face of the detached vitreous humor, which may be lined by a sheet of neovascularization (Fig. 9-2B). The term hyaloid refers to the hyaloid body, another name for the vitreous humor. Sub-ILM hemorrhages are actually hemorrhagic detachments of the ILM. Blood is located between the NFL and the outer aspect of the ILM, the retina’s single true basement membrane, which is secreted by the Müller cells. Occasionally, sub-ILM hemorrhages may be distinguished by the ophthalmoscopic observation of focal relucencies called Gunn dots on their inner surface. The latter correspond to focal concavities on the outer surface of the basement membrane that conform to the foot plates of the Müller cells.
Hemorrhagic detachment of the RPE often occurs in patients who have sub-RPE neovascular membranes. Sub-RPE hemorrhage is an important stage in the evolution of many disciform scars in age-related macular degeneration. Sub-RPE hemorrhages typically appear quite dark because blood is located beneath a layer of pigmented cells. They can be confused clinically with uveal malignant melanoma. However, in contrast to melanomas, sub-RPE hemorrhages block choroidal fluorescence and appear dark during intravenous fluorescein angiography.
Vitreous hemorrhage occurs when blood breaks through the hyaloid membrane into the substance of the formed vitreous. Blood in the formed vitreous can be quite persistent but gradually undergoes degeneration and assumes a characteristic yellow-ochre color. Nonresorbing vitreous hemorrhage is a common indication for vitrectomy, especially in patients with diabetes mellitus.
Retinal exudates and edema that occur in diabetes, hypertensive retinopathy, and other retinal vascular disorders reflect a breakdown in the blood–retinal barrier (Fig. 9-3A). The term blood–retinal barrier refers to structural modifications that protect the retina’s delicate neural tissues from fluid overload and osmotic stress. The blood–retinal barrier is analogous to the blood–brain barrier in the CNS. The inner or intraretinal part of the blood–retinal barrier is composed of the tight junctions that join together the endothelial cells that line the retinal vessels. During fluorescein angiography, these impermeable intercellular junctions confine fluorescein dye within the lumens of healthy retinal vessels. The girdle of terminal bars that join the RPE cells near their apices forms the outer part of the blood–retinal barrier. The choriocapillaris, which supplies the avascular outer third of the retina, is composed of fenestrated capillaries that leak fluorescein dye profusely. The RPE serves as the barrier that protects the outer retina from an influx of fluid.
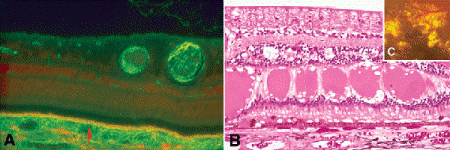
Fig. 9-3. A. Blood–retinal barrier. Tight junctions joining retinal vascular endothelial cells constitute the inner part of the blood–retinal barrier, which confines fluorescein dye within lumina of retinal vessels. Occluding junctions joining apices of RPE cells comprise outer part of barrier. Choriocapillaris is composed of leaky fenestrated capillaries. Arrow denotes yellow band of autofluorescent lipofuscin in RPE. (Freeze-dried preparation, fluorescent microscopy ×100) (From Eagle RC. Mechanisms of maculopathy. Ophthalmology 1984;91:613–625, Courtesy of Ophthalmology.) B. Retinal hard exudates. The pools of eosinophilic protein-rich fluid in the outer plexiform layer are hard exudates. Hard exudates typically occur in the OPL because that layer is the watershed zone between the retina’s two blood supplies. C. Inset shows clinical appearance of hard exudates. (B. H&E ×100)
Incompetence of either the inner or the outer part of the blood–retinal barrier gives lipid- and protein-rich fluid access to the retinal parenchyma, leading to edema and exudate formation. Hard, yellow, and waxy exudates appear histopathologically as pools of eosinophilic proteinaceous fluid (Fig. 9-3B). Hard exudates usually are located in the outer plexiform layer, the watershed zone between the retina’s dual blood supplies. In chronic cases, the exudates may be phagocytized by foamy macrophages called gitter cells.
Soft exudates or cotton-wool spots (Fig. 9-4) actually are not exudates in the true sense of the word. Cotton-wool spots represent focal areas in the NFL where the normal flow of axoplasm is blocked. This focal blockage of axoplasmic flow is thought to be a response to focal retinal ischemia and probably is caused by thrombosis of a precapillary arteriole. Cotton-wool spots are helpful clinical markers for retinal ischemia. They develop in the preproliferative phase of diabetic retinopathy and also are found in ischemic retinal vein occlusions and severe hypertensive retinopathy. Cotton-wool spots occur in relative isolation in patients who have HIV/AIDS or collagen vascular diseases such as systemic lupus erythematosus as a manifestation of intravascular immune complex deposition.
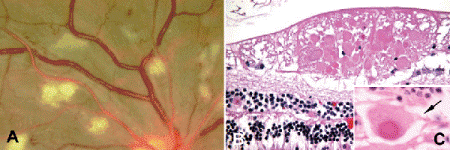
Fig. 9-4. Cotton-wool spots. A. Cotton-wool spots represent focal areas of axoplasmic flow blockage in the nerve fiber layer. They are a clinical marker for retinal ischemia. B. Histopathology of cotton-wool spot shows focus of dilated nerve fiber layer axons filled with eosinophilic axoplasm. Cytoid bodies with prominent eosinophilic nucleoids are evident. C. Cytoid body, cotton-wool spot. Eosinophilic staining distinguishes the nucleoid in the cytoid body from an actual nucleus. Arrow denotes axon entering the cytoid body. Cytoid bodies resemble cells but actually are focal areas of axonal swelling. (B. H&E ×100, C. H&E ×250)
Histopathologic examination of a cotton-wool spot shows focal swelling of the NFL and cytoid bodies (Fig. 9-4B,C). Cytoid bodies are eosinophilic segments of ganglion cell axons ballooned by stagnant axoplasm. They are called cytoid because they superficially resemble cells. Some have a nucleoid composed of aggregated organelles that mimics a cellular nucleus but is eosinophilic rather than basophilic.
ANGIOID STREAKS
Angioid streaks are linear structures seen on ophthalmoscopy that radiate from the optic disc in an angioid or vessellike fashion (Fig. 9-5). Histopathology has shown that angioid streaks correspond to breaks in Bruch membrane (Fig. 9-5B). Angioid streaks tend to develop in patients who have certain systemic disorders marked by massive calcification of Bruch membrane. Such diseases include pseudoxanthoma elasticum, Paget disease of bone, and some cases of sickle hemoglobinopathy. Calcification may impart an egg shelllike fragility to Bruch membrane predisposing to fracture. The RPE usually is intact overlying the breaks in Bruch membrane. Sub-RPE neovascularization can complicate angioid streaks and actually is a major cause of visual loss in affected patients. Unfortunately, when sub-RPE membranes develop, they generally involve the region of the macula for reasons unknown. This fact implies that the development of subretinal neovascularization requires more than a break in Bruch membrane.
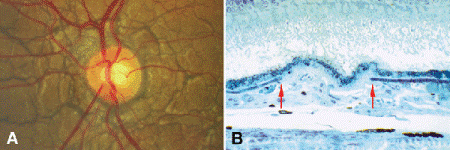
Fig. 9-5. Angioid streaks. A. The gray linear structures radiating the optic disc in an angioid or vessellike pattern are angioid streaks. They occur in several systemic disorders marked by massive calcification of Bruch membrane. B. Arrows denote margins of break in Bruch membrane. Heavily calcified Bruch membrane appears as a thick dark band beneath the RPE. The RPE is intact overlying the break in Bruch membrane. The patient had sickle cell anemia. (Toluidine blue ×150; From Jampol LM, Acheson R, Eagle RC Jr, et al. Calcification of Bruch membrane in angioid streaks with homozygous sickle cell disease. Arch Ophthalmol 1987;105:93–98, Copyright 1987, American Medical Association.)
RETINAL ARTERY AND ARTERIOLAR OCCLUSIONS
Interruption of the vascular supply to the inner two thirds of the retina causes ischemic infarction and coagulative necrosis of its cells. The sequelae of central or branch retinal artery occlusion are evident histopathologically as inner ischemic retinal atrophy (Fig. 9-6). All of the cells nourished by capillaries derived from the central retinal artery are affected in inner ischemic retinal atrophy, including the nerve fiber and ganglion cell layers, the inner plexiform layer, and most of the inner nuclear layer. The outer part of the inner nuclear layer usually persists because its cells are sustained by diffusion from the choriocapillaris. In a long-standing case, the inner retinal layers are paucicellular and have a glassy, hyalinized appearance. Gliosis of the NFL is not observed because the fibrous astrocytes and other accessory glial cells that cause NFL gliosis in chronic glaucoma perish in the retinal ictus. The additional involvement of the IPL and the inner part of the INL serves to differentiate inner ischemic retinal atrophy from glaucomatous retinal atrophy. Only the retinal ganglion cells and their axons that constitute the NFL and optic nerve are atrophic in glaucomatous retinal atrophy.
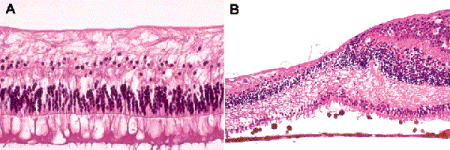
Fig. 9-6. A. Inner ischemic retinal atrophy, retinal artery occlusion. All inner retinal layers supplied by the central retinal artery are atrophic, including the nerve fiber and ganglion cell layers, the inner plexiform layer and most of the inner nuclear layer. No gliosis is present. The inner plexiform and inner nuclear layers are spared in glaucomatous retinal atrophy. B. Branch retinal artery occlusion, fovea. The fovea is seen centrally. The layers of the perfused parafoveal retina at right are well preserved. In contrast, the part of the retina supplied by the obstructed vessel (at left) shows marked inner ischemic atrophy. Photoreceptor atrophy was caused by a shallow detachment of the macula. (A. H&E ×100. B. H&E ×50)
In the acute stages of central retinal artery occlusion (CRAO), the retina shows edema, cellular dissolution, and nuclear fragmentation or pyknosis. Clinically, the normally transparent retina is marked by milky-white opacification (Fig. 9-7). A macular cherry red spot is present because the cells comprising the floor of the foveola remain viable, and therefore transparent, because they are supplied by the choriocapillaris. Although the foveal photoreceptors persist, central vision is lost because other cells in the neural pathway are destroyed. Retinal hemorrhages usually are not seen.
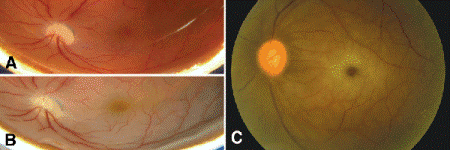
Fig. 9-7. Retinal opacification. A. The retina of freshly enucleated unfixed eye is transparent. B. Fixation causes retinal opacification (below). Cellular death caused by ischemia causes similar retinal opacification after CRAO. C. Central retinal artery occlusion. Foveola appears as cherry red spot in opacified, infarcted retina.
Occlusion of the central retinal artery produces a “ministroke” of the entire retina. Only part of the retina is infarcted in hemiretinal or branch retinal arteriolar occlusions (Fig. 9-6B). Most branch artery occlusions are caused by emboli that lodge at the bifurcation of an arteriole.
CRAO is usually a disease of elderly individuals who are often atherosclerotic, hypertensive, and diabetic. Central or branch retinal artery occlusion in a young individual should suggest the possibility of a primary cardiac tumor such as a myxoma, vasculitis, or the presence of anticardiolipin (lupus anticoagulant) antibodies. Most CRAOs result from thrombosis or embolization. Thrombosis usually occurs within the optic nerve head and is related to atherosclerosis at this site. Atherosclerosis does not develop in retinal arterioles because those vessels lack a distinct muscularis.
GIANT CELL ARTERITIS
Giant cell arteritis is an important treatable condition that must be ruled out in any elderly patient who has retinal artery occlusion or ischemic optic neuropathy. The erythrocyte sedimentation rate is typically elevated in affected individuals, who also may have thrombocytosis and elevated levels of C-reactive protein. Expedient temporal artery biopsy is performed to confirm the diagnosis. Patients who have giant cell arteritis classically have a history of malaise, weight loss, and muscle aches (polymyalgia rheumatica) and may complain of headache and painful mastication due to jaw claudication. Severely inflamed temporal arteries may be elevated, tender, and cordlike. Biopsy surgery may be relatively bloodless if the arteritis is severe.
An affected segment of artery appears firm, thickened, and opacified grossly. Histopathology discloses compromise or even occlusion of the vascular lumen and chronic inflammation within the thickened arterial wall (Fig. 9-8). The chronic granulomatous inflammatory infiltrate should contain epithelioid histiocytes, but giant cells are not particularly common in most positive biopsies, and are unnecessary for the diagnosis. Inflammation can affect all layers of the artery, but classically is concentrated in the vicinity of the internal elastic lamina, which almost invariably shows severe dissolution and segmental loss. Fibrosis and scarring are often seen in the adventitial connective tissue. Signs of old or “healed” giant cell arteritis, for example, in a patient who has received chronic corticosteroid therapy, include focal atrophy or destruction of the muscularis, extensive segmental destruction of the internal elastic lamina, and perivascular scarring. “Skip lesions” (uninflamed segments in a positive biopsy) do occur but are relatively rare. Hence, the biopsied segment of artery should be at least 2 cm long. Bilateral biopsy may be considered if the initial biopsy is negative and clinical suspicion is high, but the yield is quite low.
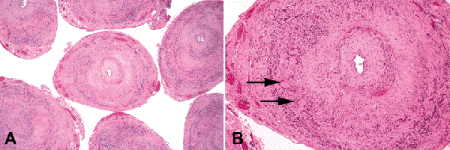
Fig. 9-8. Giant cell arteritis, temporal artery biopsy. A. The lumina of the chronically inflamed artery are largely occluded. No skip lesion are present. B. Arrows denote giant cells within chronic inflammatory infiltrate that includes epithelioid histiocytes and lymphocytes. There is extensive destruction of the lamina and the muscularis and fibrosis of the adventitia. Giant cell arteritis is an important cause of artery occlusion and ischemic optic neuropathy in elderly patients. (A. H&E ×10, B. H&E ×25)
Prompt diagnosis of giant cell arteritis is critical because bilateral blindness can develop rapidly in untreated cases. High-dose systemic corticosteroid therapy should be instituted immediately if giant cell arteritis is suspected. The temporal artery is biopsied to confirm the diagnosis histopathologically because systemic steroids can have severe side effects in elderly patients.
RETINAL VENOUS OCCLUSION
Occlusion of the central retinal vein or one of its major branches produces hemorrhagic infarction of the retina. The terms “blood and thunder fundus” or “squashed tomato sign” that have been applied clinically to this retinopathy reflect the plethora of deep and superficial retinal hemorrhages that develop after venous occlusion (Fig. 9-9A).
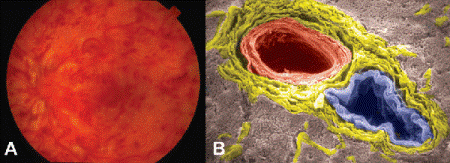
Fig. 9-9. Central retinal vein occlusion. A. Many deep and superficial hemorrhages are present. Retinal veins are dilated and tortuous. Cotton-wool spots surrounding swollen optic nerve indicate ischemia. B. Central retinal artery and vein. Common adventitial sheath (yellow) surrounds central retinal artery (red) and vein (blue) in transverse section of optic nerve. Sclerosis of the artery is a factor in the pathogenesis of many cases of CRVO. (B. False-colorized SEM ×320)
Most retinal venous occlusions involve branches of the central retinal vein, typically (70%) the superotemporal branch vein. Venous occlusions tend to occur in men who are older than age 50 years and have diabetes mellitus, hypertension, and arteriosclerosis. Local intraocular conditions that predispose to venous occlusion include elevated intraocular pressure, papilledema, and large drusen of the optic disc. Many central retinal vein occlusions are thought to be related to arteriosclerosis of the central retinal artery, which shares a common adventitial sheath with the central retinal vein within the lamina cribrosa of the optic nerve (Fig. 9-9B). The sclerotic arteriole compresses the vein within the adventitial sheath causing turbulence in the lumen of the vein, which damages the vascular endothelium and predisposes to venous thrombosis.
The early stages of a retinal venous occlusion are characterized histopathologically by diffuse and cystoid edema of the macula. The hemorrhagic retina contains numerous deep, superficial, and full-thickness retinal hemorrhages. Preretinal hemorrhages are found in some cases, and in rare instances, blood may extend into the subretinal space causing hemorrhagic retinal detachment. Other findings include shallow serous retinal detachment and papilledema. If severe ischemia and capillary nonperfusion are present, focal retinal necrosis and cotton-wool spots are noted. Cotton-wool spots are an important clinical marker for the ischemic variant of central retinal vein occlusion (CRVO).
In chronic retinal venous occlusive disease, histopathology discloses disorganization of retinal architecture and marked gliosis. The retina often contains macrophages laden with golden brown hemosiderin pigment from blood breakdown, and the retina and other epithelial structures may stain positively for iron (hemosiderosis). Inner ischemic retinal atrophy may be present if the venous occlusion is ischemic. Most eyes requiring enucleation have painful neovascular glaucoma (NVG).
Secondary closed-angle glaucoma caused by iris neovascularization (rubeosis iridis) develops in about 20% of untreated patients who have ischemic CRVOs (Fig. 8-10). Profound visual loss, cotton-wool spots, and severe nonperfusion of the retinal capillary bed disclosed by fluorescein angiography are clinical signs of ischemic vein occlusion. This type of glaucoma has been called “90-day glaucoma,” reflecting its fairly rapid onset. In the past, most of these blind, painful glaucomatous eyes were enucleated. Today, modern therapies including panretinal photocoagulation and tube shunts have dramatically diminished the number of eyes enucleated after CRVO.
HYPERTENSIVE RETINOPATHY
The retinopathy that develops in patients with severe systemic hypertension is caused by vascular incompetence and breakdown of the blood–retinal barrier. Acute severe elevation of blood pressure causes retinal arteriolar narrowing and focal vasospasm. If elevated blood pressure levels and vasospasm persist chronically, the muscular and endothelial coats of the vessels eventually become necrotic. Histopathologic studies have revealed changes in the endothelial lining, necrosis of the smooth muscle, and insudation of fibrin-rich plasma in the vessel wall. The endothelial damage causes vascular incompetence with resultant retinal edema, exudation, and occasionally even serous retinal detachment. Small exudates called edema residues may form a stellate pattern around the fovea (macular star figure). This pattern of exudation is governed by the radial orientation of the photoreceptor axons (Henle fibers) in the perifoveal outer plexiform layer (OPL). In the early days of ophthalmoscopy, severe hypertensive retinopathy with a macular star figure was often called hyperalbuminuric retinitis, reflecting the common association between severe hypertension and renal failure. Retinal hemorrhages and papilledema are additional manifestations of hypertensive retinopathy. Retinal hemorrhages are relatively common and occur in the disorder’s early stages. Optic disc edema marks the fourth and final stage of hypertensive retinopathy, is an important clinical marker for malignant hypertension and potential encephalopathy, and an indication for aggressive antihypertensive therapy as well. Fibrinoid necrosis caused by the insudation and accumulation of plasma proteins in vessel walls may affect retinal and choroidal vessels. Occlusion of small, damaged vessels also occurs, causing microinfarctions of the NFL (cotton-wool spots), and occasionally, infarctions of larger areas of retina. Focal choroidal infarction with pigmentary change may be evident clinically as Elschnig spots and Siegrist streaks. These were considered to be grave prognostic signs before effective antihypertensive therapy became available. Retinal macroaneurysms occasionally develop in hypertensive patients.
RETINAL ARTERIOSCLEROSIS
Chronic low-grade hypertension induces fibrosis in the walls of retinal arterioles, a process called retinal arteriolarsclerosis (Fig. 9-10). Histopathologically, the sclerotic retinal vessels are encompassed by a thick mantle of collagenous connective tissue. The term “onion skin” is often applied to this change.
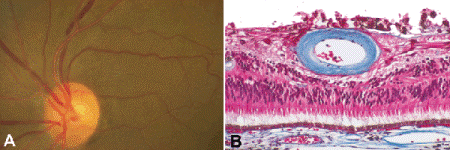
Fig. 9-10. Retinal arteriolarsclerosis. A. Fundus photo shows copper-hued sclerotic arterioles and A-V crossing defects. B. Thick mantle of collagenous connective tissue surrounds sclerotic vessel. Blue staining with Masson trichrome (below) confirms the presence of collagen. (B. Masson trichrome ×100)
Like the surrounding neurosensory retina, the walls of healthy retinal vessels normally are transparent. What one observes ophthalmoscopically as retinal vessels actually are the columns of pigmented erythrocytes filling the lumina of the vessels. The progressive accumulation of connective tissue in the vessel walls of patients with retinal arteriosclerosis gradually obscures the blood column, widening the light reflex and imparting an orange or coppery hue to the arterioles (Fig. 9-10A). Eventually, if the process is prolonged and severe, perivascular fibrosis may totally hide the blood column, and the vessels will appear as white lines or silver wires. Arteriovenous crossing defects (a-v nicking) result when the opaque walls of thickened arterioles obscure part of the underlying venules. In advanced cases, there may be deflection or banking of the retinal vein.
Cystoid macular edema (CME) is characterized by the accumulation of serous fluid in cystoid spaces in the parenchyma of the perifoveal retina (Fig. 9-11). The intraretinal cysts have a characteristic petalloid appearance on intravenous fluorescein angiography, and are disclosed by optical coherence tomography (OCT). This pattern undoubtedly reflects the radial orientation of the Henle fibers in the perifoveal OPL. Fine has suggested that CME initially may begin as intracellular edema in Müller cells. Cystoid spaces presumably form as a consequence of cellular death in the milieu of chronic edema. Histopathologically, the cystoid spaces appear relatively empty or contain scant amounts of granular or fibrinous material. The latter serves to distinguish them from hard exudates, which are usually pools of eosinophilic hyaline material.
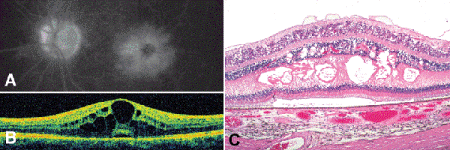
Fig. 9-11. Cystoid macular edema. A. IVFA discloses petalloid pattern of intraretinal cystoid edema in fovea. B. OCT graphically shows cysts and retinal thickening. C. Large cystoid spaces filled with granular proteinaceous fluid are seen in the outer plexiform and inner nuclear layers. Thickened retina is detached artifactitiously. (C. H&E ×50)
Inflammatory mediators such as prostaglandins that are made in the anterior segment are responsible for some cases of CME. The dramatic response of CME to antivascular endothelial growth factor therapy indicates that vascular endothelial growth factor (VEGF) also plays an important pathogenic role. Vitreous traction on the macula has been incriminated in other instances. Visual loss due to CME may be the presenting clinical manifestation of peripheral lesions including tumors or peripheral uveitis (pars planitis). CME is an important complication of ocular surgery. The association of CME with cataract surgery is called the Irvine-Gass syndrome. A relatively high incidence of CME occurred in patients who had iris-supported intraocular lenses (IOLs) implanted after intracapsular cataract surgery. The IOL probably stimulated prostaglandin production by the iris, and complete removal of the lens allowed the inflammatory mediator to readily diffuse to the posterior segment. CME can complicate any severe chronic ocular inflammatory disorder. Visual loss may respond to medical therapy in early cases.
AGE-RELATED MACULOPATHY (AMD, AGE-RELATED MACULAR DEGENERATION, ARMD)
Age-related maculopathy (AMD) or age-related macular degeneration (ARMD) is the leading cause of irreversible blindness in people 50 years of age or older in the developed world. (Legal blindness generally is defined as a best-corrected visual acuity of 20/200 or 6/60.) AMD causes loss of central vision because it involves the fovea, the specialized part of the retina used for high-resolution color vision (Figs. 9-12–9-14). Although affected patients have trouble reading and recognizing faces, they retain their peripheral vision and are able to ambulate. Anxious patients may be assured that they will never go totally blind. (This in not entirely true, however, since patients receiving anticoagulant therapy occasionally develop massive hemorrhagic retinal detachment.)
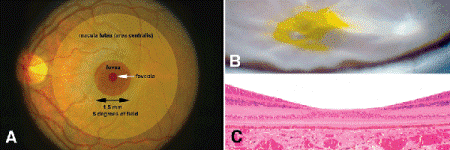
Fig. 9-12. A. The “macula.” The foveola is located in the center of the 1.5-mm-wide fovea or pit. The area centralis is delimited by the temporal vascular arcades. The imprecise term macula (from macula lutea, “yellow spot”) is often applied clinically to this region. B. Irregular yellow spot of carotenoid xanthophyll pigment that encompasses the fovea is readily seen in opacified retina after fixation. C. Retina comprising foveal floor consists only of photoreceptors, outer nuclear layer, and part of outer plexiform layer. (C. H&E ×25)
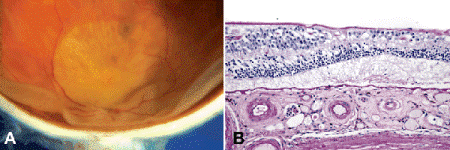
Fig. 9-13. Atrophic form of age-related macular degeneration. A. Extensive area of RPE atrophy involves posterior pole in eye removed from a nonagenarian with a long history of blindness who underwent enucleation for invasive mucoepidermoid carcinoma. Window defect reveals choroidal vessels. B. Corresponding histopathology shows total loss of RPE, photoreceptors, and outer nuclear layer. Outer retina is fused to Bruch membrane, which is thickened and PAS positive. The choriocapillaris has undergone involution. (B. PAS ×50)
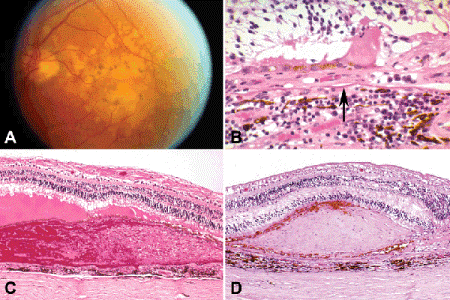
Fig. 9-14. Exudative form of age-related macular degeneration. A. Large confluent soft drusen blanket posterior retina. B. Subretinal neovascular membrane. A thin layer of fibrous tissue containing capillaries elevates the atrophic RPE from the inner surface of Bruch membrane. The atrophic outer retina shows extensive photoreceptor loss. Bruch membrane (arrow) is fractured and focally calcified. The choroid contains an incidental focus of chronic inflammatory cells. C. The retinal pigment epithelium is detached by blood and vascularized connective tissue. A collagenous disciform scar is forming on the inner surface of Bruch membrane as the hemorrhagic RPE detachment undergoes organization. D. Disciform scar. Mound of collagen incorporating RPE and vessels elevates fovea. Severe photoreceptor atrophy is present. (B. H&E ×100, C. H&E ×25, D. H&E ×25)
There are atrophic or dry and exudative or wet forms of AMD. The simultaneous or sequential development of dry and wet changes in a single patient suggests that both are variants of a clinical and pathologic spectrum. Dry or atrophic macular degeneration is characterized by atrophy and death of subfoveal RPE, which leads to photoreceptor degeneration, outer retinal atrophy, and involution of the choriocapillaris (Fig. 9-13).
Yellowish white subretinal deposits called drusen are clinical hallmarks of AMD (Figs. 9-14A and 9-15). Drusen are focal deposits of extracellular debris located between the basal lamina of the RPE and the inner collagenous layer of Bruch membrane. The composition of drusen is complex. Constituents are numerous and include neutral lipids, unesterified cholesterol, carbohydrates, vitronectin, C-reactive protein, apolipoprotein E, and a variety of proteins involved in inflammation such as amyloid-beta, immunoglobulin light chains, factor X, complement factor H, C5, and the C5b complex.
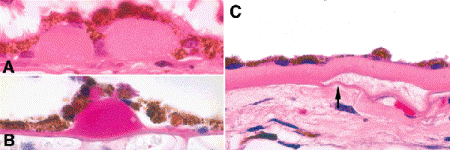
Fig. 9-15. Drusen. A. Hard drusen. Hard drusen are excrescences of homogeneous hyaline material on the inner surface of Bruch membrane that focally detach the RPE. B. Many hard drusen are intensely PAS positive. C. Basal laminar deposit (diffuse soft drusen). A thick band of abnormal basement membrane material elevates the retinal pigment epithelium off the inner surface of Bruch membrane. Part of the deposit has detached artifactitiously from Bruch membrane (arrow). The RPE cells are flattened and atrophic. Basal laminar deposits predispose to RPE detachment and age-related macular degeneration. (A. H&E ×250, B. PAS ×250, C. H&E ×250)
Several varieties of drusen are recognized clinically, and a number of somewhat confusing and occasionally conflicting classification schemes based on histopathologic and ultrastructural features have been proposed. Hard or cuticular drusen are most abundant and are found throughout the retina. They are discrete, round or globular mounds of homogeneous, deeply periodic acid-Schiff (PAS)-positive hyaline material (Fig. 9-15A,B). Soft drusen are found only in the region of the macula and are strongly associated with AMD. Soft drusen typically comprise loose amorphous material called membranous debris. Their contents may be liquefied and often is lost during histologic processing. Soft drusen are often associated with diffuse deposits of extracellular matrix material called basal laminar deposits. Basal laminar deposits play an extremely important role in the pathogenesis of AMD, especially the severe exudative type, which is complicated by the formation of subretinal neovascular membranes and disciform scar formation.
Basal laminar deposits appear light microscopically as extensive plaques or layers of “soft” granular eosinophilic material that elevate the atrophic RPE from the inner surface of Bruch membrane (Fig. 9-15C). Transmission electron microscopy has shown that such deposits are located between the plasma membrane and the basement membrane of the RPE and are composed of extracellular matrix material rich in “curly collagen” or 1,000 Å banded basement membrane material. Basal laminar deposits adhere loosely to Bruch membrane, predisposing to RPE detachments and tears. The deposits theoretically could interfere with biochemical modulation of the choriocapillaris by the RPE and also could provide a plane for sub-RPE neovascular invasion. A second less important variety of diffuse soft drusen called a basal linear deposit has been identified electron microscopically. Basal linear deposits are located within Bruch membrane external to the RPE basement membrane and are composed of multivesicular particles of lipoprotein, which are also the primary constituent of soft drusen. It is impossible to distinguish these two varieties of diffuse soft drusen with routine light microscopy.
Subretinal neovascular membranes characterize the exudative type of AMD (Fig. 9-14B). Angiogenic factors, most notably VEGF, stimulate the proliferation of the new vessels. Clinically, patients typically present with decreased visual acuity or distorted vision (“metamorphopsia”). Membranes appear as grayish patches on ophthalmoscopy, frequently with associated hemorrhage and overlying subretinal fluid. Intravenous fluorescein angiography is used to confirm the presence of neovascularization. OCT also can disclose the neovascular membranes but is used primarily to evaluate retinal thickness and cystoid edema and gauge response to anti-VEGF therapy. In the past, laser photocoagulation was used to obliterate the vessels. In recent years, however, laser therapy has been largely supplanted by the intravitreal injection of monoclonal antibodies or antibody fragments such as bevacizumab or ranibizumab that target VEGF. Such agents are highly efficacious in many cases, but they must be administered repeatedly by intravitreal injection at relatively frequent intervals. Neovascularization and its complications recur if therapy is stopped. Anti-VEGF therapy halts the progression of AMD and improves vision in many patients but does not address the underlying causes of the disease.
In untreated cases, the new vessels leak or bleed forming serous and/or hemorrhagic detachments of the RPE (Fig. 9-14C). When the RPE detaches, the plane of the detachment usually is between the inner surface of Bruch membrane and a basal laminar deposit, which detaches with the RPE. If new vessels have invaded a basal laminar deposit, they are apt to be sheared off during detachment, causing hemorrhagic detachment of the RPE. Fibrous disciform scar formation, the endstage of exudative AMD, may result from the organization of hemorrhagic RPE detachment. Histopathologically, mature scars are composed of mounds of dense collagenous connective tissue on the inner surface of Bruch membrane (Fig. 9-14D). The collagenous scar usually contains vessels and aggregates of RPE cells. The outer part of the overlying retina undergoes degeneration because the disciform scar is a solid retinal detachment that separates the photoreceptors from their usual source of nourishment. The collagenous part of the scar is derived in part from the fibroblastic component of the granulation tissue that invades and organizes the sub-RPE hemorrhage. The RPE also contributes to scar formation. RPE cells are able to produce large quantities of extracellular matrix material including drusenoid basement membrane material, collagen, and even bone.
Heredity plays a role in the susceptibility to AMD, but development of the disorder appears to depend on a complex interplay of genetic and environmental factors. Environmental risk factors include a history of smoking, white ethnicity, obesity, high dietary intake of vegetable fat, and low dietary intake of antioxidants and zinc. Other contributing factors appear to be photooxidative damage, the accumulation of lipofuscin and chromophores that accumulate within and damage RPE cells, accumulation of lipid in Bruch membrane that impedes its conductivity, and hypoxia caused by involution of choriocapillaris.
There is recent evidence that suggests that chronic inflammation may play an important role in the pathogenesis of AMD. Cellular remnants and debris from damaged RPE cells trapped between the RPE and Bruch membrane are thought to trigger local up-regulation of cytokines, acute phase reactants such as C-reactive protein, and inflammatory mediators causing activation of the complement cascade. The debris also may attract choroidal dendritic cells that function as antigen-presenting cells. Immunohistochemical studies have disclosed complement components C5 and complement attack complex C5b-9 in drusen and damaged RPE cells. This observation is quite important because patients who have a characteristic polymorphism in the gene for complement factor H (CFH) are at significantly greater risk for AMD. CFH encodes the major inhibitor of the complement alternative pathway.
Subretinal neovascular membranes also complicate other conditions such as angioid streaks. Idiopathic subretinal membranes occasionally occur in relatively young individuals without antecedent cause. Ocular histoplasmosis syndrome (OHS) typically affects patients from the Ohio Valley or other areas where histoplasmosis is endemic (Fig. 9-16). Histopathologically, the disciform scars found in patients with OHS resemble those seen in AMD but typically have a prominent infiltrate of lymphocytes in the underlying choroid (Fig. 9-16B). In addition to disciform macular scars, patients with OHS have peripapillary chorioretinal atrophy and multiple white “punched-out” chorioretinal scars (Fig. 9-16A). The latter also contain chronic inflammatory cells.
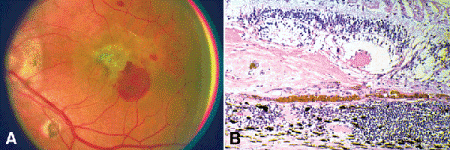
Fig. 9-16. Ocular histoplasmosis syndrome. A. Fundus photo shows triad of hemorrhagic disciform macular scar, peripapillary chorioretinal atrophy, and “punched-out” chorioretinal scars. B. Disciform scar, ocular histoplasmosis syndrome. A mound of connective tissue containing vessels and RPE cells rests on Bruch membrane and focally detaches the retina. The photoreceptors are atrophic. Chronic inflammation is seen in the underlying choroid. (B. H&E ×100)
THE RETINAL PIGMENT EPITHELIUM
The RPE is vital to the health and survival of the overlying retina (Fig. 1-4). Responsible for most of the characteristic reddish brown color of the human fundus, the RPE absorbs excess light and prevents intraocular light scattering like the black coating on the inside of a camera. The RPE is also involved in the transport of fluid, nutrients, and vital metabolites to and from the outer retina, and the regeneration of the visual pigment rhodopsin. It also appears to play a major active role in retinal adherence by actively pumping fluid from the subretinal space. Finally, the RPE phagocytizes and digests the tips of millions of damaged and discarded photoreceptor outer segments every day.
STARGARDT DISEASE (FUNDUS FLAVIMACULATUS)
Striking RPE abnormalities occur in several inherited human retinal diseases including Stargardt disease or fundus flavimaculatus (Fig. 9-17). Affected patients lose vision in their teens from an atrophic type of macular degeneration caused by the death of the subfoveal RPE. The term fundus flavimaculatus, applied to this disorder by Franceschetti, literally means “yellow-spotted fundus” and refers to characteristic yellow pisciform spots at the level of the RPE disclosed by ophthalmoscopy. The fundus may have a vermilion hue, and a striking clinical abnormality called the dark choroid or the sign of choroidal silence is evident on intravenous fluorescein angiography. This obscuration of the normal choroidal pattern is caused by the massive accumulation of an abnormal lipofuscin-like lipopigment in the cytoplasm of the RPE cells. The surfeit of pigment absorbs excitatory blue wavelengths during angiography and makes the RPE intensely PAS positive (Fig. 9-17B,C). The RPE cells are taller than normal, and their nuclei are often displaced toward the apex of the cell. Scanning electron microscopy (SEM) has revealed markedly enlarged RPE cells, which are more numerous in the posterior part of the fundus (Fig. 9-17A). Groups of abnormal enlarged RPE cells surrounded by smaller relatively normal cells may be responsible for the yellow-flecked appearance of the fundus. All of the RPE cells contain yellow-brown lipopigment, but the lipofuscin is hidden by a relatively normal complement of apical melanin in the smaller cells. The pisciform aggregates of larger cells appear yellow because these grossly abnormal cells are relatively amelanotic. Macrophages or detached, lipopigment-laden RPE in the subretinal space may also contribute to the flecked retinal appearance. The massive accumulation of pigment probably contributes to RPE dysfunction and death. The death of subfoveal RPE cells in turn leads to photoreceptor degeneration and atrophic macular degeneration.
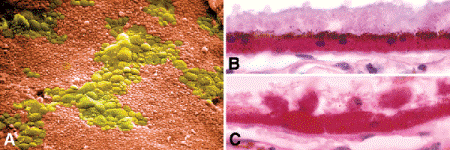
Fig. 9-17. Stargardt disease (fundus flavimaculatus). A. RPE abnormalities. False-colorized scanning electron micrograph shows aggregates of markedly enlarged RPE cells in posterior part of eye obtained postmortem from a patient with autosomal recessive fundus flavimaculatus. Hypothetically, the groups of larger cells may appear as yellow spots clinically because they have less apical melanin pigment than the surrounding smaller, more normal cells. B. RPE cells are packed with granules of PAS-positive material consistent with an abnormal form of lipofuscin. Nuclei in small peripheral RPE cells are displaced apically. C. Posterior RPE cells contain more lipofuscin and are abnormally large, amelanotic, and focally detached. The abnormal pigment in the RPE blocks normal choroidal fluorescence during intravenous fluorescein angiography, producing a dark choroid. The massive accumulation of lipofuscin is caused by a toxic vitamin A derivative contained in ingested photoreceptor outer segments. The ABCA4 gene is expressed only in rod outer segments. (A. False-colorized SEM ×120 [From Eagle RC et al. Retinal pigment epithelial abnormalities in fundus flavimaculatus: a light and electron microscopic study. Ophthalmology; 1980;87:1189–1200. Courtesy of Ophthalmology], B. PAS ×250, C. PAS ×250)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


