15 Optic Nerve
DEVELOPMENTAL ANOMALIES
Developmental anomalies of the optic nerve include optic nerve aplasia and hypoplasia, optic pits, optic nerve colobomas, and the morning glory syndrome. Bilateral hypoplasia is often associated with congenital syndromes such as de Morsier syndrome of septo-optic dysplasia, which includes bilateral hypoplastic nerves, absent septum pellucidum, and hemiplegia, and Aicardi syndrome, which affects only women and includes peripapillary chorioretinal lacunae, ectopic retinal pigment epithelium (RPE), agenesis of the corpus callosum, infantile spasms, and mental retardation.
Colobomas of the optic nerve (Fig. 15-1) are caused by incomplete closure of the posterior portion of the fetal fissure. Eyes with extensive optic nerve colobomas may be microophthalmic and have a cystic out-pouching of the posterior sclera (microphthalmos with cyst) (Fig. 2-2). The cyst typically is lined by dysplastic neuroectodermal tissue, which communicates with the retina via the coloboma. Optic nerve colobomas occasionally are associated with choristomatous malformations that contain smooth muscle and heterotopic fat.
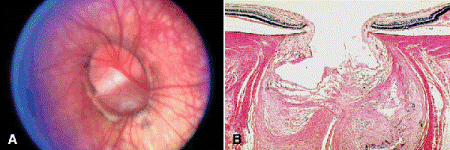
Fig. 15-1. A, B. Optic nerve coloboma. Optic nerve colobomas are caused by incomplete closure of the fetal fissure. (B. H&E ×10)
Optic pits are small craterlike holes that usually occur unilaterally at the temporal margin of the optic disc. Pathogenesis probably is related to anomalous closure of the superior margin of the embryonic fissure. Optic pits frequently are complicated by serous detachment of the macula, complicated by schisis of its inner layers. The subretinal fluid may be derived from the vitreous.
The morning glory syndrome is an optic nerve anomaly characterized by a funnel-shaped optic nerve head, which appears to contain a central dot of connective tissue believed to be residual Bergmeister papilla. The retinal vessels emerge from the margin of the disc, which is surrounded by an elevated annulus of disturbed chorioretinal pigment. Bilateral cases occasionally have been associated with midline neurologic and craniofacial anomalies.
OPTIC DISC DRUSEN
Optic disc drusen are globular aggregates of concentrically laminated, calcified material that are located deep in the substance of the optic nerve head anterior to the lamina cribrosa within the scleral ring (Fig. 15-2). Optic disc drusen appear ophthalmoscopically as tan, yellow, or straw-colored glistening or refractile spheric structures. They typically are found in a small, crowded optic disc that has a small or absent cup. Optic disc drusen are unrelated to drusen of the RPE or the heavily calcified epipapillary astrocytomas called giant drusen of the optic disc that occur in some patients with tuberous sclerosis complex. Optic disc drusen are important clinically because they may be misdiagnosed as papilledema and prompt an unnecessary neurologic evaluation. The pathogenesis of optic disc drusen may be related to blockage of axoplasmic flow in ganglion cell axons within a narrow crowded scleral canal. Calcified mitochondria dispersed from prelaminar corpora amylacea may provide a nidus for further calcium deposition. Optic disc drusen occur sporadically or may be inherited as an irregular autosomal dominant trait. Disc drusen also occur in some patients with retinitis pigmentosa or pseudoxanthoma elasticum with angioid streaks.
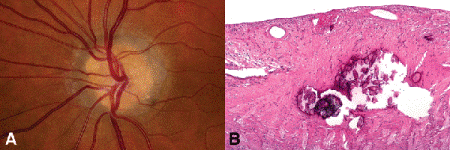
Fig. 15-2. Optic disc drusen. A. Anterior substance of optic disc contains yellow spherical refractile bodies. Optic disc drusen may be misdiagnosed clinically as papilledema. B. A conglomeration of calcareous deposits is present in the optic nerve anterior to the lamina cribrosa. The drusen have fractured during sectioning. They were unsuspected, and the specimen was not decalcified. (A. Photo courtesy of Dr. Peter Savino, B. H&E ×25)
Although optic disc edema (papilledema) classically is associated with elevated intracranial pressure and space-occupying intracranial lesions, swollen optic discs also occur in eyes with acute glaucoma, ocular hypotony, central retinal vein occlusion, juxtapapillary tumors, and severe hypertensive retinopathy (Fig. 15-3). Optic disc edema does not result from an accumulation of fluid in the extracellular spaces of the disc. Rather, the increase in the volume of the nerve head reflects the intracytoplasmic swelling and thickening of ganglion cell axons caused by blockage of axoplasmic flow in the distorted lamina cribrosa. The pores of the lamina cribrosa are distorted by a pressure gradient between the intraocular pressure and pressure in the retrolaminar optic nerve. A pressure gradient can form if the intracranial pressure is elevated (classic papilledema), the intraocular pressure is low (hypotony), or the intraocular pressure is acutely elevated (acute glaucoma).
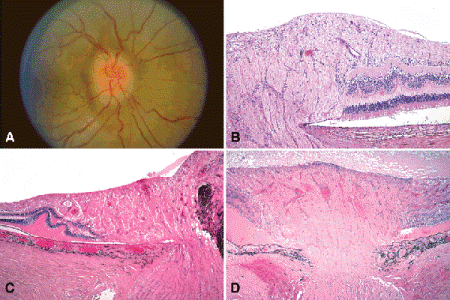
Fig. 15-3. Optic disc edema. A. The optic nerve is swollen and injected and has blurred margins. Concentric folds are seen in the adjacent retina. B. The nerve head is swollen. The photoreceptors are displaced laterally. C. Optic disc edema, secondary to juxtapapillary melanoma. The optic nerve head is compressed by an infiltrating juxtapapillary tumor, causing blockage of axoplasmic flow. The photoreceptors of the swollen optic nerve are displaced laterally and the peripapillary retina is detached by serous fluid. Folds are noted in the outer retina. D. Optic disc edema, hypotony. The optic disc is massively swollen. The photoreceptors are displaced laterally, and a shallow exudative retinal detachment is present. Severe hypotony caused by uveitis was the cause of the disc edema. (B. H&E ×50, C. H&E ×25, D. H&E ×25)
Histopathologically, the nerve head is swollen and the physiological cup is narrowed (Fig. 15-3B–C). The increase in the volume of nerve head tissue displaces the photoreceptors laterally from the margin of the disc. This lateral displacement of photoreceptors and an accompanying shallow peripapillary collection of serous subretinal fluid are responsible for enlargement of the blind spot on visual field testing. Folds are also found in the outer retinal layers (Paton folds). Extensive gliosis and axonal loss occur in chronic papilledema.
OPTIC ATROPHY
Optic nerve atrophy is characterized pathologically by shrinkage of the parenchyma of the optic nerve caused by loss of ganglion cell axons (Fig. 15-4). The subarachnoid space around the shrunken nerve becomes widened, and the dura may appear redundant and folded. Light microscopy discloses loss of axons and thickening of the pia mater and pial septa. Gliosis may or may not become prominent depending on the cause of the atrophy.
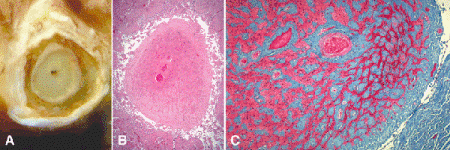
Fig. 15-4. Optic atrophy. A. The optic nerve is atrophic, and the subarachnoid space is widened. The severity of the optic atrophy makes the meninges appear redundant. B. The pia and pial septa are markedly thickened, and the substance of the nerve is severely atrophic. C. Transverse section shows that most of the parenchyma of the severely atrophic optic nerve has been replaced by blue-staining collagenous connective tissue. The pia and pial septa are markedly widened. (B. H&E ×10, C. Masson trichrome ×25)
By convention, the terms primary or descending optic atrophy are applied to atrophy of the nerve caused by lesions in the central nervous system or orbit. Primary optic atrophy generally is not associated with an ophthalmoscopically visible glial or mesenchymal reaction. Causes of primary optic atrophy include optic nerve trauma, compression by neoplasms or enlarged extraocular muscles in thyroid ophthalmopathy, neurosyphilis, demyelinating diseases including multiple sclerosis, heritable leukodystrophies, and toxic and nutritional optic neuropathies.
Inflammatory, neoplastic, or vascular lesions located in the retina or the vicinity of the optic disc cause secondary or ascending optic atrophy, which is often marked by pronounced alterations in the glial and mesenchymal tissues of the nerve head. Common retinal causes of optic atrophy include chorioretinitis, retinitis pigmentosa, and trauma.
Leber hereditary optic neuropathy is caused by several point mutations in genes encoding complex I subunits of the mitochondrial respiratory chain. The subunit 4 gene of mitochondrial DNA encoding NADH dehydrogenase is mutated most often. Mitochondrial DNA is maternally inherited. Leber hereditary optic atrophy usually presents with subacute progressive bilateral central visual loss in men between 18 and 30 years of age. Some patients have disc swelling and telangiectatic peripapillary vessels.
Although retinal ganglion cells and axons are lost in both primary and glaucomatous optic atrophy, cupping of the disc generally occurs in glaucomatous optic atrophy and is not prominent in primary optic atrophy. Schnabel’s cavernous optic atrophy is a relatively rare type of optic atrophy characterized by the presence of large spaces filled with hyaluronic acid in the retrolaminar part of the nerve (Fig. 15-5). Schnabel’s cavernous optic atrophy classically was associated with an acute elevation of intraocular pressure, but a large postmortem study found many cases in elderly women with systemic vascular disease who had no evidence of glaucoma. Hypothetical sources of the mucopolysaccharide include the vitreous in glaucomatous eyes or in situ production within areas of optic nerve infarction. No gliosis or histiocytic reaction typically is seen. Intraocular silicone oil can infiltrate the optic nerve producing pseudo-Schnabel’s cavernous degeneration. In rare instances, the oil can migrate to the brain.

Fig. 15-5. Schnabel’s cavernous optic atrophy. A. Diameter of transversely sectioned optic nerve is markedly widening. Small cystoid spaces replace myelinated parenchyma. B. Retrolaminar optic nerve contains pools of clear mucoid material. C. Clear spaces in retrolaminar optic nerve stain intensely for acid mucopolysaccharide (AMP). D. Positive staining is abolished by pretreatment with hyaluronidase indicating that the substance is hyaluronic acid. (B. H&E ×10, C. Colloidal iron for AMP ×10, D. colloidal iron after hyaluronidase digestion ×10)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


