12 Retinoblastoma and Simulating Lesions
Retinoblastoma is the most common intraocular tumor of childhood. In Asia, Africa, and South America, where uveal melanoma is relatively rare, it probably is the most common primary intraocular tumor. Retinoblastoma is relatively rare; only 300 cases occur yearly in the United States. The incidence has been estimated to be 1 in 15,000 to 1 in 34,000 births. All races and both sexes are affected equally, and the tumor has no predilection for either the right or left eye. The mean age at diagnosis is 18 months, and about 90% of cases are diagnosed before 3 years of age. Rare cases in older children and adults are often misdiagnosed.
CLINICAL PRESENTATION
In the United States and Europe, about 90% of patients present with leukocoria, an abnormal, typically white pupillary reflex that has been fancifully likened to the tapetal light reflex of the cat (amaurotic cat’s-eye reflex) (Fig. 12-1). The white pupillary reflex is caused by tumor in the vitreous cavity (endophytic tumors) or the detached retina (exophytic tumors). Strabismus occurs in about one third of cases. For this reason, careful ophthalmoscopy should be performed on all children with strabismus to exclude retinoblastoma or some other significant retinal pathology. Rarer presentations include neovascular glaucoma (NVG), which may cause secondary buphthalmos and iris heterochromia. Some eyes with endophytic or diffuse infultrative retinoblastomas develop pseudohyppyons of tumor cells in the anterior chamber. Aseptic orbital cellulitis caused by servere NVG that induces intraocular necrosis is another pseudoinflammatory presentation. Patients in underdeveloped countries often present in the late stages of the disease with proptosis or an orbital mass caused by extraocular extension of the tumor (Fig. 12-2). Exceptional cases of clinically manifest congenital retinoblastoma have been reported. These present with a massive hyphema and an enlarged ectatic cornea that spontaneously perforates. It has been estimated that half of retinoblastomas actually may be present at birth but are inapparent clinically.
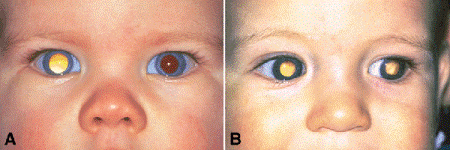
Fig. 12-1. A. Leukocoria. Unilateral sporadic retinoblastoma. About 90% of patients with retinoblastoma in the United States present with a white pupillary reflex. B. Bilateral leukocoria, familial retinoblastoma. Bilateral tumors occur in about two thirds of patients with familial retinoblastoma. The presence of bilateral tumors indicates that the affected patient is a carrier of familial retinoblastoma who can transmit the tumor to progeny. (Photos courtesy of Dr. Jerry A. Shields, Wills Eye Hospital.)
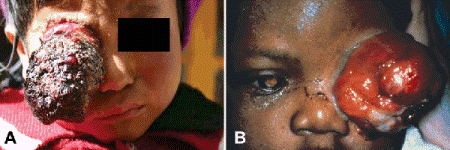
Fig. 12-2. A. Tibetan child with massive orbital involvement by retinoblastoma. Children in underdeveloped countries who have little access to medical care often present with in the late stages of the disease with proptosis or an orbital mass. B. Neglected case of bilateral retinoblastoma from the United States. Tumor fills left orbit. Leukocoria heralds a large tumor in the fellow eye. (From Zimmerman LE. Retinoblastoma, including a report of illustrative cases. Med Ann Dist Columb 1969;38:366–374.)
GROSS PATHOLOGY AND GROWTH PATTERNS
Macroscopically, retinoblastoma has a white encephaloid or brainlike appearance, which is not surprising since the tumor arises from the retina, a peripheral colony of brain cells (Fig. 12-3). Lighter flecks of calcification or necrotic tumor usually are evident grossly in the tumor. Necrotic retinoblastomas found in infants with aseptic orbital cellulitis typically have a blood-tinged, orange, or soupy, grayish necrotic appearance.
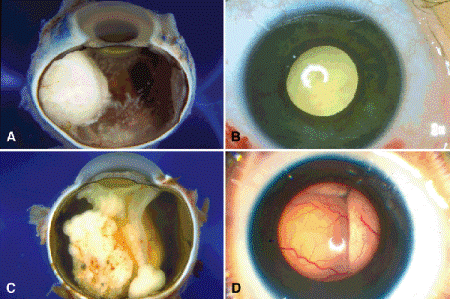
Fig. 12-3. Growth patterns. A. Endophytic retinoblastoma. Encephaloid tumor arises from inner retina, which remains attached. Extensive vitreous seeding is present. B. Leukocoria is caused by tumor in vitreous cavity. C. Exophytic retinoblastoma. Exophytic retinoblastomas arise from the outer layers of the retina and cause retinal detachment. Macrophoto of enucleated eye with exophytic retinoblastoma shows encephaloid tumor in subretinal space and total bullous retinal detachment, which adheres to the back of the lens. The lens-iris diaphragm is displaced anteriorly causing secondary closure of the angle. D. Retinal vessels are visible behind lens in eyes with exophytic retinoblastoma.
Tumors with endophytic, exophytic, mixed or indeterminate, and diffuse infiltrating growth patterns occur. Endophytic retinoblastomas arise from the inner layers of the retina, which remains attached. The tumor invades the vitreous cavity and can seed the anterior chamber (Fig. 12-3A,B). A pseudohypopyon of tumor cells can develop if there is extensive seeding of the anterior chamber. Hence, endophytic tumors can be confused with primary inflammatory disorders such as toxocariasis, mycotic endophthalmitis, or granulomatous uveitis.
Exophytic retinoblastomas arise from the outer layers of the retina and cause retinal detachment (Fig. 12-3B,C). The detached retina is often highly elevated, and its vessels are visible behind the lens on clinical examination. Exophytic retinoblastomas are usually confused clinically with simulating lesions such as Coats disease that cause an exudative retinal detachment. Totally endo- or exophytic retinoblastomas are actually relatively uncommon; most tumors have a mixed endophytic-exophytic or indeterminate growth pattern. About 1.4% of retinoblastomas diffusely thicken the retina without forming a distinct mass (Fig. 12-4). This rare diffuse infiltrating growth pattern usually is found in older children (mean age 6 years) who typically present with pseudoinflammatory signs and invariably have unilateral sporadic tumors. During gross or histopathologic examination, it is nearly impossible to distinguish multifocal primary lesions spawned by germline mutations from secondary tumors that result from tumor seeding.
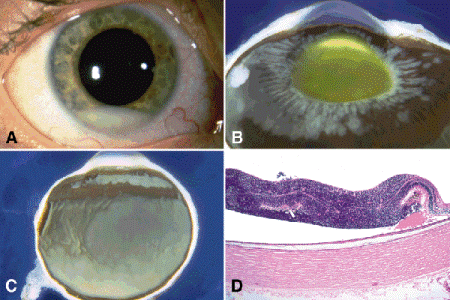
Fig. 12-4. Diffuse infiltrating retinoblastoma. A. Pseudohypopyon. Layered deposit of tumor cells in quiet eye was presenting manifestation of diffuse infiltrating retinoblastoma in older child. B. Infiltration of zonule by tumor occurs in many eyes with diffuse infiltrating tumors. C. The neoplasm diffusely infiltrates, thickens, and opacifies part of the retina but does not form a distinct mass. Tumor is present on the pars plana. The unilateral sporadic tumor was found in a 7-year-old boy who presented with anterior chamber seeding. D. The tumor diffusely infiltrates and thickens the retina but does not form a distinct mass. Calcification is absent. (D. H&E ×10)
HISTOPATHOLOGY
Under low magnification, retinoblastoma appears as a basophilic mass with pink and purple foci that arises from and destroys the retina and fills part or all of the vitreous cavity (Fig. 12-5). The basophilic areas are composed of viable retinoblastoma cells. The poorly differentiated neuroblastic cells appear blue because they have intensely basophilic nuclei and scanty cytoplasm (Fig. 12-5A). Numerous mitoses and fragments of apoptotic nuclear debris usually are present. Retinoblastoma grows rapidly and has a marked propensity to outgrow its blood supply and undergo spontaneous coagulative necrosis. This usually occurs when the proliferating cells have extended about 90 to 110 μm away from a blood vessel (Fig. 12-5D). The necrotic tumor cells lose their basophilic nuclear DNA and become pink or eosinophilic. The residual viable cells typically form cuffs or sleeves around vessels, imparting a multilobulated or papillary appearance to some tumors (Fig. 12-5C,D). These perivascular cuffs were called pseudorosettes by some in the past. Foci of dystrophic calcification develop in the necrotic parts of the tumor in many cases (Fig. 12-6). Histopathologically, the calcific foci appear reddish purple in hematoxylin and eosin sections, and the presence of calcium can be confirmed by the von Kossa or alizarin red stains. Electron microscopy suggests that calcification probably begins in the mitochondria of necrotic cells. Clinically, the demonstration of calcification by ultrasonography or computed tomography can help to differentiate retinoblastoma from other simulating lesions.
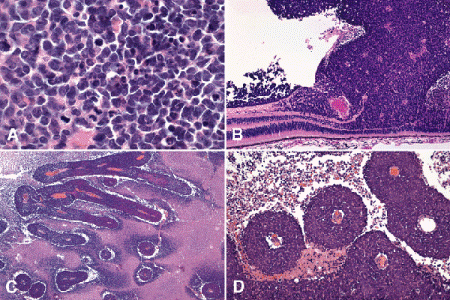
Fig. 12-5. Histopathology. A. Viable parts of a retinoblastoma appear blue in H&E stained sections because the tumor is composed of poorly differentiated neuroblastic cells that have scanty cytoplasm and prominent basophilic nuclei. Mitoses and apoptotic cells are common. B. Endophytic retinoblastoma. This poorly differentiated basophilic neoplasm arises from and destroys the retina. The retina remains attached as the endophytic tumor invades the vitreous cavity. Rosettes are not seen. C. Low magnification photomicrograph of retinoblastoma showing basophilic areas of viable tumor and eosinophilic zones of necrosis. The viable cells form characteristic sleeves and cuffs around vessels. D. Perivascular cuffs of tumor cells. Retinoblastoma typically outgrows its blood supply and undergoes necrosis. The necrotic cells become eosinophilic because they lose their nuclear DNA. Necrosis generally occurs when its cells have grown approximately 90 to 100 μm away from a vessel. The persistent viable cells form basophilic sleeves and cuffs around vessels. (A. H&E ×250, B. H&E ×50, C. H&E ×10, D. H&E ×50)
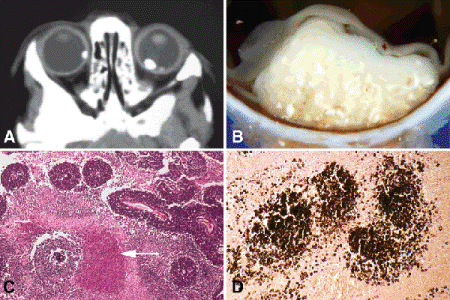
Fig. 12-6. Calcification. A. CT scan shows foci of residual calcified tumor in both eyes after chemoreduction therapy. B. Calcium and necrosis are evident grossly as lighter flecks in tumor. C. Dystrophic calcification develops in necrotic parts of tumor. The calcific foci appear purple in H&E stained sections (arrow). D. Calcified nuclei of tumor cells stain positively with von Kossa stain for calcium. (A. Photo courtesy of Carol L. Shields, MD, Wills Eye Institute. C. H&E ×25, D. von Kossa ×100)
Intensely basophilic deposits of DNA released from necrotic tumor cells are another characteristic histopathologic feature of retinoblastoma (Fig. 12-7). DNA deposition generally is found in eyes with extensively necrotic tumors. Typically, the DNA deposits surround retinal or iris vessels or are found in the trabecular meshwork or in basement membranes such as the lens capsule or the internal limiting membrane of the retina.
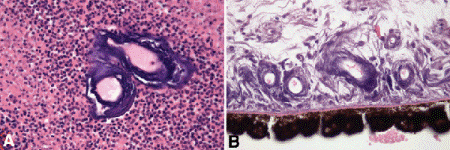
Fig. 12-7. DNA deposition. A. Basophilic material surrounding vessels is nuclear DNA released by necrotic cells. The surrounding tumor cells are necrotic. B. DNA deposition in iris stroma. DNA also may deposit in the ILM and the wall of the Schlemm canal. (A. H&E ×100, B. H&E ×100)
Retinoblastoma cells often collect on the inner surface of the Bruch membrane, causing focal retinal pigment epithelium (RPE) detachments. Such sub-RPE deposits of tumor cells do not constitute choroidal invasion. During histopathologic examination, care must be taken to distinguish artifactual contamination of the choroid (and epibulbar tissues) from true invasion. The presence of a mixture of viable and necrotic cells is one feature that serves to identify artifactual seeding. Foci of extramedullary hematopoiesis also can be confused with uveal invasion in rare instances.
More than 40% of eyes enucleated for retinoblastoma have iris neovascularization, which may produce NVG, iris heterochromia, and even secondary buphthalmos. NVG is almost three times more common in eyes with high-risk features such as massive choroidal invasion or retrolaminar optic nerve invasion.
TUMOR DIFFERENTIATION: ROSETTES AND FLEURETTES
Varying degrees of retinal differentiation occur in retinoblastoma. This is evident as Homer Wright and Flexner-Wintersteiner rosettes and photoreceptor differentiation (fleurettes) as well (Figs. 12-8 and 12-9). Flexner-Wintersteiner rosettes represent an early attempt at retinal differentiation. Histologically, these rosettes are composed of a ring of cuboidal cells surrounding a central lumen (Fig. 12-8B). The lumen, which corresponds to the subretinal space, contains hyaluronidase-resistant acid mucopolysaccharide (AMP) similar to photoreceptor matrix AMP. The cells surrounding the lumen are joined near their apices by intercellular connections (zonulae adherentes), analogous to the external limiting membrane of the retina. Cilia, which exhibit the 9 + 0 pattern of microtubular doublets found in the central nervous system, project into the lumen. Cilia are hypothesized to be the precursor of photoreceptor outer segments. Although highly characteristic of retinoblastoma, Flexner-Wintersteiner rosettes are not pathognomonic because they do occur in malignant medulloepitheliomas and some pineal tumors.
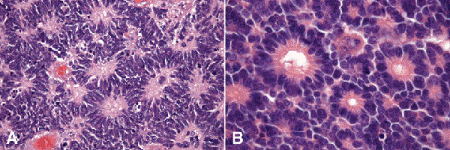
Fig. 12-8. A. Homer Wright rosettes. Each ring of cells surrounds a central tangle of neural processes. A central lumen is not present. Homer Wright rosettes are indicative of neuroblastic differentiation. They are the least differentiated form of rosette found in retinoblastoma and also occur in other tumors such as neuroblastoma. B. Flexner-Wintersteiner rosettes. Flexner-Wintersteiner rosettes represent differentiation toward primitive retina. The central lumen corresponds to the subretinal space. The tumor cells surrounding the lumen are joined near their apices by cellular connections that are analogous to the external limiting membrane. (A. H&E ×250, B. H&E ×250)
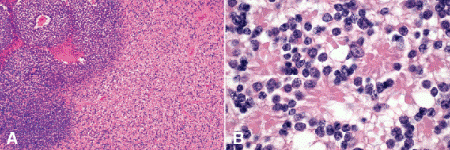
Fig. 12-9. Retinoblastoma, photoreceptor differentiation. A. Photoreceptor differentiation typically is found in an area of viable tumor that appears relatively eosinophilic compared to poorly differentiated retinoblastoma. B. Neoplastic photoreceptors typically form bouquets of pink “flowers” called fleurettes. The inner segments of the neoplastic photoreceptors appear as bulbous eosinophilic processes that often are aligned along a segment of neoplastic external limiting membrane. The nuclei are bland and mitoses and necrosis are not evident. Tumors composed entirely of fleurettes are considered to be benign variants of retinoblastoma called retinomas or retinocytomas. (B. H&E ×250)
Homer Wright rosettes (named after James Homer Wright) indicate neuroblastic differentiation (Fig. 12-8A). They lack a central lumen, and their constituent cells encompass a central tangle of neural filaments. Wright rosettes are relatively nonspecific. They also occur in neuroblastoma and are a characteristic feature of cerebellar medulloblastoma.
Retinoblastoma tends to become less well-differentiated with age. Numerous Flexner-Wintersteiner rosettes typically are found in eyes enucleated from younger infants, while tumors in older children tend to be poorly differentiated. A recent study found that the mean age at enucleation for tumors with many rosettes was 10.4 months; for moderate rosettes, 18.3 months; for sparse rosettes, 20.4 months; and for poorly differentiated tumors, 33.9 months. These differences were statistically significant. The presence of many Flexner-Wintersteiner rosettes in a tumor may have prognostic significance. In one series, patients who had moderately well-differentiated tumors that contained abundant Flexner-Wintersteiner rosettes had about a sixfold better prognosis than those whose tumors lacked rosettes.
About 15% to 20% of retinoblastomas harbor very well-differentiated foci of actual photoreceptor differentiation (Fig. 12-9). Such areas contain aggregates of neoplastic photoreceptors called fleurettes by Ts’o, Zimmerman, and Fine, who described them in 1970. Photoreceptor differentiation typically is found in areas of viable tumor that appear relatively eosinophilic and paucicellular compared to adjacent undifferentiated retinoblastoma on low-magnification microscopy of H&E stained sections (Fig. 12-8A). The term “fleurette” denotes a bouquetlike arrangement of cytologically benign cells joined by a series of zonulae adherentes comprising a short segment of neoplastic external limiting membrane. Neoplastic photoreceptor inner segments evident as bulbous eosinophilic processes form the “flowers” of the bouquet (Fig. 12-8B). Electron microscopy has disclosed stacks of cellular membranes representing early outer segment differentiation in some cases. The demonstration and characterization of photoreceptor differentiation firmly established that retinoblastoma was not a retinal glioma and affirmed that the adoption of the name retinoblastoma by the American Ophthalmological Society in 1926 at Frederick Verhoeff’s suggestion was indeed appropriate.
A retinal tumor composed entirely of photoreceptor differentiation is now thought to be a benign variant of retinoblastoma called a retinoma or retinocytoma. The cells comprising retinoma/retinocytoma are quite bland compared to those of retinoblastoma with a low nuclear cytoplasmic ratio, finely dispersed chromatin, and no apoptosis, mitoses, or necrosis. Calcification is found but occurs in viable parts of the tumor.
Retinomas or retinocytomas initially were thought to represent spontaneously regressed retinoblastomas on clinical grounds because they resemble retinoblastomas that have regressed after radiation therapy (Fig. 12-10A). They have a translucent “fish flesh” appearance, contain abundant calcification that has been likened to cottage cheese, and are surrounded by a ring of RPE depigmentation. Retinomas/retinocytomas generally are small tumors that are found in eyes that retain useful vision. They may be found incidentally or are discovered in a parent or sibling when the detection of retinoblastoma in a child prompts examination of other family members. Retinocytomas are relatively resistant to radiation, as are other benign tumors. Therefore, it is not surprising that foci of photoreceptor differentiation are found more often in eyes that are enucleated after external-beam radiotherapy or chemotherapy.
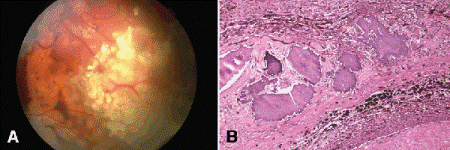
Fig. 12-10. A. Retinocytoma. The tumor has a translucent “fish flesh” appearance and contains large amounts of calcification that has been likened to cottage cheese. An annulus of RPE depigmentation typically surrounds the tumor. The tumor shown here was detected at a preschool vision screening and was observed unchanged for several years before undergoing malignant transformation. (From Eagle RC Jr, et al. Malignant transformation of spontaneously regressed retinoblastoma, retinoma/retinocytoma variant. Ophthalmology 1989;96:1389–1395. Courtesy of Ophthalmology.) B. Spontaneously regressed retinoblastoma, phthisical eye. Basophilic foci of necrotic calcified tumor cells are the only remnants of retinoblastoma in this phthisical eye. The calcified tumor is encased by scar tissue that contains strands of hyperplastic RPE. The choroid is scarred and shows evidence of necrosis. (B. H&E ×25)
NATURAL HISTORY AND PROGNOSTIC FACTORS
Retinoblastoma is a highly malignant neoplasm that grows relentlessly and is invariably fatal if untreated. The tumor arises from the retina and invades the vitreous cavity and/or the subretinal space. Tumor cells eventually breach the Bruch membrane and invade the choroidal stroma (Fig. 12-11A–D). Choroidal vessels serve as a major route for distant hematogenous dissemination, and massive choroidal invasion is an indication for adjuvant chemotherapy in many centers. Involvement of the anterior chamber, iris stromal, and trabecular meshwork also are thought to affect prognosis adversely, but the actual magnitude of this effect is uncertain. Of 297 previously untreated eyes enucleated for retinoblastoma at the Wills Eye Institute, 16.5% were found to have uveal invasion, which was classified as massive (>3 mm in diameter) in about half.
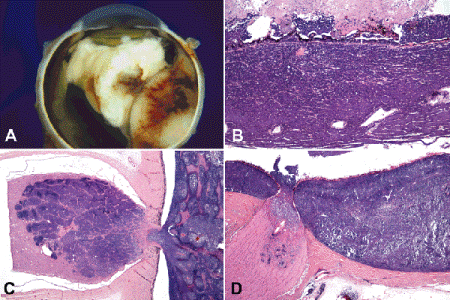
Fig. 12-11. High-risk histopathologic findings. A. Retinoblastomas, massive choroidal invasion. The right half of the choroid is massively thickened by invasive retinoblastoma. The area of choroidal invasion contains foci of hemorrhage. Careful examination disclosed no extraocular extension. The patient developed metastases after the parents declined prophylactic chemotherapy. B. Retinoblastoma, massive choroidal invasion. The entire thickness of the choroid is replaced by a basophilic infiltrate of poorly differentiated, viable retinoblastoma cells. Massive choroidal invasion recently has been defined as a full-thickness infiltrate >3 mm in diameter. C. Retinoblastoma, optic nerve invasion. There is massive replacement of the retrolaminar parenchyma of the optic nerve by basophilic tumor. Tumor is seen in close proximity to the cut surface of the nerve. A cross section of nerve containing the true surgical margin was excised and submitted for sections before the eye was opened. Equivalent to extraocular extension, retrolaminar optic nerve invasion is an indication for adjuvant chemotherapy in many centers. D. Eye with concurrent massive choroidal invasion and retrolaminar invasion of optic nerve. Multiple high-risk features often occur together. (B. H&E ×50, C. H&E ×25, D. H&E ×10)
Retinoblastoma has a marked proclivity to invade the optic nerve, and optic nerve invasion is an extremely important prognostic factor (Fig. 12-11C,D). The tumor can travel along the optic nerve to the brain, or malignant cells may be dispersed along the neuraxis if they gain access to the cerebrospinal fluid. Of the 297 untreated eyes in the Wills Eye Institute series, 38.7% had some degree of optic nerve invasion. Retrolaminar optic nerve invasion, an important indication for adjuvant chemotherapy, was present in about 10.4%. Mortality rates correlate directly with the depth of optic nerve invasion. In one series, 10% died if there was superficial invasion of the nerve head only (grade I) and 29% if the tumor reached and invaded the lamina cribrosa (stage II). Mortality rose to 42% when there was retrolaminar invasion (grade III) and 78% when the tumor extended to the surgical margin (grade IV). Hence, surgeons should try to obtain as long a segment of optic nerve as possible when enucleating an eye that is known or suspected to harbor retinoblastoma. In addition, enucleation should not be performed by an inexperienced surgeon.
After retinoblastoma has filled the globe and destroyed its contents, it extends extraocularly. Anteriorly, the tumor extends through the aqueous outflow pathways, preexisting emissarial canals, or perforations in the cornea. Posterior segment tumors that have invaded the choroid can extend extraocularly through emissarial canals or can invade the orbit by directly infiltrating and destroying the sclera. Secondary buphthalmos and staphylomas caused by secondary glaucoma can facilitate extrascleral extension. Secondary closed-angle glaucoma caused by pupillary block or iris neovascularization is relatively common in eyes with retinoblastoma. About 43% of eyes enucleated for retinoblastoma have iris neovascularization and 26% have NVG. In addition, NVG is much more common in eyes with high-risk histopathologic features.
Retinoblastoma typically metastasizes hematogenously to lungs, bones, brain, and other organs. Cervical and preauricular adenopathy can develop when tumors with extensive anterior segment involvement gain access to lymphatics in the conjunctival stroma. Lymphatics are not present in the orbit. Metastatic disease usually becomes evident within 1 or 2 years after therapy. Late metastasis is so rare in retinoblastoma that it should raise the possibility of a second independent primary tumor such as pineoblastoma.
Retinoblastoma occasionally undergoes spontaneous regression. A typical bona fide example of spontaneous regression is a phthisical eye that contains foci of calcified retinoblastoma cells. Both the tumor regression and phthisis bulbi probably are caused by extensive ischemic necrosis in an eye with severe NVG (Fig. 12-10B). Many lesions that previously were thought to be spontaneously regressed tumors actually are retinomas or retinocytomas.
MOLECULAR GENETICS AND THE RETINOBLASTOMA ONCOGENE
Retinoblastoma is a hereditary cancer: 5% to 10% of the tumors are inherited in what appears to be a classic Mendelian autosomal dominant trait; carriers transmit the tumor to one half of their offspring (Fig. 12-12B). Bilaterality is a characteristic feature of heritable retinoblastomas. Bilateral tumors occur in about 60% of patients who have germline mutations in the RB1 gene. The average age of patients with familial retinoblastoma is about 1 year. A germline mutation should be suspected when retinoblastoma is found in a very young infant.
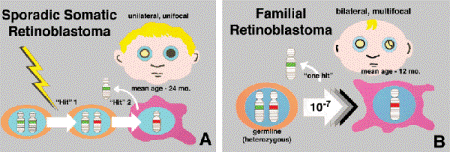
Fig. 12-12. A. Sporadic somatic retinoblastoma. Retinoblastomas caused by sporadic somatic mutations are most common. They are always unilateral and typically occur at about age 2 years. B. Familial retinoblastomas typically are bilateral and multifocal. They occur earlier than tumors caused by sporadic somatic mutations because only a single “hit” or gene inactivation is required.
The great majority of retinoblastomas (85%) are sporadic tumors that arise in patients with a negative family history (Fig. 12-12A). About 75% of these sporadic tumors are caused by somatic mutations in retinal cells. Sporadic retinoblastomas caused by somatic mutations are invariably unilateral, unifocal tumors that cannot be passed-on to progeny, and they tend to occur in older infants (mean age 2 years). The remaining 25% of sporadic cases are caused by germline mutations and represent new familial cases that are transmissible.
A small number (<5%) of retinoblastomas occur in infants who have a variety of congenital anomalies and are found to have deletions in the long arm of chromosome 13 that are evident in karyotypic analysis. In addition to retinoblastoma, the 13Q- deletion syndrome comprises severe mental retardation and other anomalies including microcephaly, hypertelorism, ptosis, micrognathia, deformed low-set ears, a wide nasal bridge, cardiac anomalies, anal atresia, microphthalmia, colobomas, and cataracts. The association of retinoblastoma with the 13Q-syndrome initially suggested that the retinoblastoma gene was located on chromosome 13.
The paradigmatic human recessive oncogene or tumor suppressor gene, the retinoblastoma or RB1 gene is located in the 14 band of the Q or long arm of chromosome 13 (13q14). The RB1 gene is 180,388 base pairs in length, and its protein product pRB comprises 928 amino acids. pRB is abundant in the nucleus, where it is involved in control of the cell cycle. During the G1 or resting phase of the cell cycle, pRB is bound to transcription factors such as E2F. Phosphorylation of pRB causes release of E2F. Uncomplexed E2F, in turn, activates a variety of other genes and transcription factors that are important in the initiation of DNA synthesis (S phase). Absence of pRB causes continual cell division and lack of terminal differentiation. The RB protein is phosphorylated by cyclin D2 and its cyclin-dependent kinase (cdk) partner. Certain oncoviruses cause tumors by synthesizing proteins (e.g., adenoviral protein E1A and SV40 large T protein) that bind to pRB and inactivate it.
Classically, the Rb1 gene is thought to cause cancer when its protein product is absent or dysfunctional (Fig. 12-13). Healthy persons have two normal or wild-type Rb1 genes (Fig. 12-13A). Both alleles of the Rb1 gene are absent or inactivated in the retinoblastoma cells. Carriers of familial retinoblastoma are heterozygous for the Rb1 gene. Although the pRB produced by a heterozygote’s single functional gene is sufficient to inhibit tumorigenesis, heterozygotes are at substantial risk to develop retinoblastoma. Although retinoblastoma appears to be inherited clinically as an autosomal dominant trait, the gene is recessive at a molecular level (Fig. 12-14).
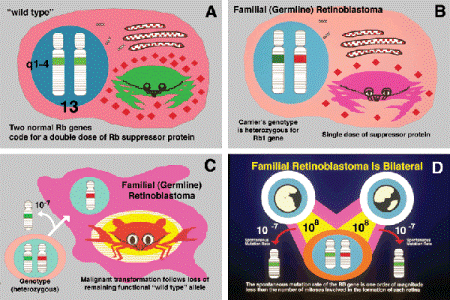
Fig. 12-13. The retinoblastoma gene. A. Normal individuals have two copies of the retinoblastoma gene, which is located on the long arm of chromosome 13. The gene encodes Rb protein that suppresses tumor formation. B. Patients with familial retinoblastoma are heterozygous for the retinoblastoma gene. C. Retinoblastoma develops when both copies of the retinoblastoma gene in a retinal cell are lost or inactivated. D. The spontaneous mutation rate of the Rb gene is less that the number of mitoses required for the formation of each retina. This is the basis for bilateral and multifocal tumors in heterozygous carriers.
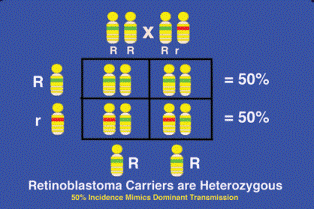
Fig. 12-14. Familial retinoblastoma appears to be an autosomal dominant trait, but the gene actually is recessive at the molecular level.
The genotype of a child with familial retinoblastoma includes one functional copy of the Rb1 gene (Fig. 12-13B). The second copy of the gene is mutated and encodes dysfunctional RB protein or it may be absent. Retinoblastoma develops when the solitary remaining wild-type gene is lost or inactivated, by chance, in a cell within the developing retina (Fig. 12-13C). Cytogenetic mechanisms responsible for gene inactivation (and resultant homozygosity for the recessive allele) include chromosomal loss or deletion, somatic recombination, and point mutation. The spontaneous mutation rate of the normal wild-type Rb1 gene is 1 in 10 million or greater. It is estimated that 100 million mitoses occur during the growth and development of each retina. Hence, it is highly probable that the second functional copy of the Rb1 gene will be lost or inactivated in at least one retinal cell in a carrier with a heterozygous genotype. Furthermore, it is equally probable that gene inactivation and tumorigenesis will occur in both eyes (Fig. 12-13D).
Immune surveillance appears to arrest some tumors, despite the appropriate additional mutations. This probably is responsible, in part, for the incomplete penetrance of the Rb1 gene, which is estimated to be approximately 80% (in other words, there is an 80% chance that one tumor will develop in one eye). A few families with low-penetrance retinoblastoma have been reported. They have reduced levels of wild-type RB protein or mutant RB protein that retains partial activity.
About one third of patients with retinoblastoma have bilateral tumors (Figs. 12-1B and 12-15). The sporadic occurrence of bilateral tumors indicates that the affected patient has a germline mutation and is capable of transmitting the disease to one half of his or her offspring. Unfortunately, the opposite is not true. As a result of incomplete penetrance and expressivity, 10% to 15% of unilateral, sporadic retinoblastomas are caused by potentially transmissible, germline mutations. Statistics used for genetic counseling (Table 12-1) reflect both the effect of gene penetrance and the proportion of familial, chromosomal deletion, and sporadic somatic and germline retinoblastomas in the population.
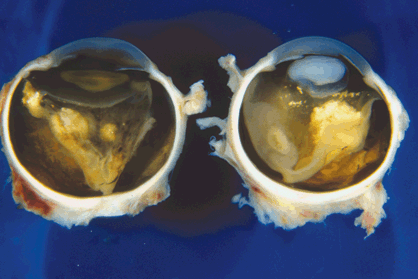
Fig. 12-15. Bilateral retinoblastoma. Both eyes had to be enucleated after therapy failed to control the tumors. Bilateral involvement indicates that the patient has a potentially transmissible germ-line mutation in the Rb gene.
TABLE 12-1 Genetic Counseling: Risk that Subsequent Child Will Have Retinoblastoma

Molecular genetics readily explains several hitherto puzzling clinical features of retinoblastoma. For example, retinoblastoma is predominantly a tumor of early childhood. Although rare adult cases have been reported, the mean age at diagnosis is 18 months and the tumor is extremely rare after age 4 years. Cytogenetic misadventures that cause gene inactivation generally occur during cellular division. Most mitotic activity in the retina actually ceases before birth, making neoplastic transformation in older persons highly unlikely. Retinoblastomas caused by germ-line mutations typically develop at an earlier age than retinoblastomas caused by sporadic somatic mutations (mean age 12 months vs. 23 months), presumably because only a single Rb1 gene allele, rather than two, must be inactivated. Knudson graphically compared the ages of patients who had unilateral and bilateral tumors with the logarithm of the proportion in each group as yet undiagnosed. His results led him to postulate that two separate events or “hits” are necessary for the development of sporadic retinoblastomas (“two-hit hypothesis”). The curve for bilateral, hereditary tumors is a simple exponential relation, consistent with a single gene inactivation or “hit” superimposed on the inherited genotypic defect.
Some of the most interesting secondary nonocular neoplasms that develop in patients with germline mutations are tumors of the pineal gland or parasellar region that resemble ectopic retinoblastomas. This association between pinealoma (also termed pineoblastoma) and bilateral hereditary retinoblastoma has been termed “trilateral retinoblastoma.” The pineal gland, which serves as a “third eye” in some primitive reptiles, shares antigenic determinants with the retina and exhibits transient photoreceptor differentiation in the neonatal rat. Photoreceptor differentiation has been identified in human pineal tumors.
The Rb1 gene has been implicated as a contributing factor in a variety of other systemic malignancies, including breast, lung, and bladder cancer. This is not surprising, considering the Rb1 gene’s fundamental role in control of the cell cycle.
Recent molecular genetic studies suggest that the initial concepts outlined above concerning the Rb1 tumor suppressor gene and its role in the pathogenesis of retinoblastoma are an oversimplification. Dimaras recently has shown that both copies of the Rb1 gene are inactivated and Rb protein is absent in retinoma/retinocytoma, which is now considered to be a benign precursor lesion of retinoblastoma. Additional mutations are necessary for malignant transformation of retinoma into retinoblastoma. The retinoblastoma gene plays an important role in the regulation of cellular division in all cells in the body, yet only retinoblastoma, a relatively rare neoplasm that affects a small population of highly differentiated cells, results from the inactivation of both copies of the RB1 gene. The highly retinal-specific predisposition imposed by mutations in the RB1 gene is hypothesized to result from the specific pattern of expression of other genes in the unidentified cell of origin in the developing human retina rather than RB1.
THE CLASSIFICATION AND TREATMENT OF RETINBLASTOMA
There are several staging systems for retinoblastoma. The older Reese-Ellsworth classification has prognostic significance for retention of an eye, maintenance of sight, and the control of local disease; but is complicated; and recently has become less useful because it is based on the response to external beam radiotherapy, a treatment modality that has fallen out of favor and is now used infrequently. The newer International Classification of Retinoblastoma (ICRB) is a relatively simple, practical classification based on clinical findings that was designed primarily to evaluate the potential response to modern therapy including chemoreduction. The International Classification includes five stages A through E. Tumors with localized subretinal seeds are placed in group C and more diffuse seeding in group D. Group E tumors general require enucleation. They fill more than 50% of the globe or have opaque media, NVG, or high risk features such as postlaminar optic nerve invasion, significant choroidal invasion, and invasion of the anterior chamber, sclera, or orbit.
A detailed discussion of the treatment of retinoblastoma is beyond the scope of this chapter. A number of reports dealing with modern treatment modalities are referenced below. In short, there have been major changes in the treatment of retinoblastoma in developed countries in recent years. A concerted effort has been made to avoid the use of external beam radiotherapy, which leads to disfiguring facial deformities and predisposes retinoblastoma gene carriers to secondary malignant neoplasms in the field of radiation such as soft tissue sarcomas. Instead, many infants with retinoblastoma, particularly those with germline mutations and bilateral tumors are being treated with chemotherapy. In many instances, chemotherapy is used to shrink tumors so they are amenable to additional treatment or “consolidation” with local therapeutic agents such as cryotherapy, infrared laser transpupillary thermotherapy, or radioactive plaque brachyradiotherapy. Called chemoreduction, this form of chemotherapy is used most commonly in the treatment of bilateral retinoblastoma. Many eyes with unilateral retinoblastoma are still enucleated. A few centers are treating retinoblastoma by direct infusion of chemotherapy into the ophthalmic artery. This controversial experimental technique directly targets the tumor and may decrease complications of systemic chemotherapy.
In developed countries, retinoblastoma is one of the great success stories of pediatric ocular oncology. At some centers, survival rates from the primary tumor approach 100%, and secondary tumors are now the primary cause of death. Unfortunately, this is not the case in underdeveloped countries, where patients frequently present with advanced stages of the disease and therapy generally is palliative (Fig. 12-2).
THE DIFFERENTIAL DIAGNOSIS OF RETINOBLASTOMA
Retinoblastoma must be differentiated clinically from a variety of benign childhood disorders that cause leukocoria. The three most common conditions that are confused with retinoblastoma are ocular toxocariasis, persistent hyperplastic primary vitreous (PHPV), and Coats disease (see Table 12-2).
TABLE 12-2 The Differential Diagnosis of Retinoblastoma
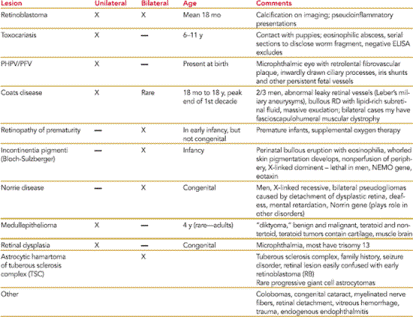
Ocular toxocariasis, particularly the nematode endophthalmitis form of the disorder, is the most common inflammatory disease that simulates retinoblastoma (Fig. 12-16A). Ocular toxocariasis is a localized ocular manifestation of visceral larva migrans caused by second-stage larvae of the canine ascarid Toxocara canis. Ocular toxocariasis usually is unilateral and occurs in children toward the end of the first decade. Diffuse nematode endophthalmitis, retinal detachment, or an eosinophilic abscess located anteriorly in the vitreous can cause leukocoria. Subfoveal granulomas occasionally present with visual loss in eyes with clear media. Histopathology discloses necrotic larval fragments surrounded by granulomatous inflammation containing many eosinophils (Fig. 12-16A). Serial sections may be necessary to demonstrate the parasite in a presumptive eosinophilic abscess. Clinically, an ELISA test for Toxocara antigen excludes the diagnosis if it is negative. A positive ELISA test does not exclude retinoblastoma, however, because antibodies to T. canis are common in some populations.
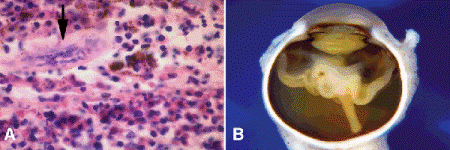
Fig. 12-16. A. Ocular toxocariasis. Arrow points to fragment of nematode in eosinophilic abscess. B. Incontinentia pigmenti. Enucleated eye has total retinal detachment caused by florid vitreoretinal neovascularization. Fellow eye had nonperfusion of the peripheral retina. Patient had characteristic dermal pigmentation. (A. H&E, ×100)
Other inflammatory diseases that occasionally are misdiagnosed as retinoblastoma include endogneous endophthalmitis, congenital toxoplasmosis, cytomegalovirus retinitis, herpes simplex virus retinitis, and peripheral uveoretinitis.
PERSISTENT HYPERPLASTIC PRIMARY VITREOUS/PERSISTENT FETAL VASCULATURE (PHPV/PFV)
PHPV/PFV is an extremely common variety of pseudoretino- blastoma (Fig. 12-17). A congenital anomaly, PHPV/PFV is present at birth. Although exceptions have been reported, PHPV/PFV classically occurs in a microphthalmic eye that has a shallow anterior chamber and a clear lens. The disorder usually is unilateral. Histopathologically, a plaque of fibrovascular tissue that resembles primary vitreous adheres to the posterior lens capsule, and the hyaloid artery is often patent. The tips of the ciliary processes typically adhere to the margins of the retrolenticular plaque. As the eye enlarges, the processes are drawn centrally and elongated. Visible behind the clear lens through a dilated pupil, these inwardly drawn ciliary processes are a helpful diagnostic sign (Fig. 12-17A). Abnormal shunt vessels often traverse the iridic surface, and other parts of the fetal vasculature may persist.
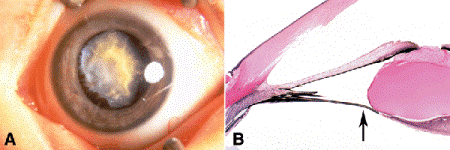
Fig. 12-17. Persistent hyperplastic primary vitreous. A. Inwardly drawn ciliary processes adhere to the margin of vascularized retrolental fibrous plaque. The lesion was present at birth and the affected eye was microphthalmic. B. Long attenuated ciliary process (arrow) attaches to margin of fibrous plaque on posterior surface of lens. An anterior subcapsular cataract also is present. (B. H&E ×5)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


