Ptosis Surgery
Aaron Savar
Sean M. Blaydon
Tanuj Nakra
John W. Shore
Blepharoptosis or ptosis refers to a congenital or acquired descent of the upper eyelid below physiologic levels.1 Ptosis can be classified by age of onset or by etiology. The age of the ptosis patient is an important factor in management as ptosis in infants or children may result in or contribute to the development of amblyopia. Ptosis in the young patient requires prompt diagnosis, monitoring, and treatment, if necessary. Ptosis may also cause visual loss in the adult by obstruction of the superior visual field.2 Although ptosis is a common condition, its diagnosis and treatment can be challenging. Proper classification of ptosis during examination lays the foundation for further workup, as well as for planning management options. The two general categories of ptosis, congenital and acquired, are further subclassified by etiology (e.g., aponeurotic, neurogenic, myogenic, mechanical, and traumatic). In this chapter, we discuss the various types of ptosis, and we present subjective and objective cues to aid in diagnosis, evaluation, and treatment.
CONGENITAL PTOSIS
Congenital ptosis is a condition present from birth and is unilateral in 75% of cases.3 Children with ptosis require careful assessment of amblyopia risk. Amblyopia occurs in 20% of congenital ptosis patients; only 4% of that amblyopia is attributed to the ptotic eyelid occluding the pupil. Other causes of ptosis-associated amblyopia include anisometropia, high astigmatism, or strabismus.2 In all cases, early assessment and treatment of ptosis is necessary to prevent permanent vision loss when the visual axis is partially or completely obstructed. If the patient already demonstrates significant amblyopia, there is an extremely high rate of ptosis surgery failure. Functional vision is required to generate frontalis muscle tone, which may be recruited during surgery for these patients. Amblyopia should be addressed in conjunction with ptosis surgery.
Congenital Myogenic Ptosis
The most common type of congenital ptosis is myogenic ptosis. The pathophysiology involves dysgenesis of the levator palpebrae superioris (LPS) muscle. The skeletal muscle fibers of the LPS are replaced by fibroadipose tissue (Fig. 1). This causes decreased levator muscle function resulting in ptosis. Both elevation and relaxation of the eyelid are impaired in congenital ptosis: Lagophthalmos on downgaze is the sine qui non of congenital ptosis and is caused by the fibrosis of the LPS, which restricts relaxation of the muscle. If the levator tone is markedly reduced or absent, there may be absence of an eyelid crease (Fig. 2). The upper eyelid crease is caused by pretarsal LPS-dermis attachments, and patients with congenital ptosis may not have enough LPS tone to produce an eyelid crease. Lash ptosis may also be seen in myogenic congenital ptosis from decreased tone and loss of structural support.
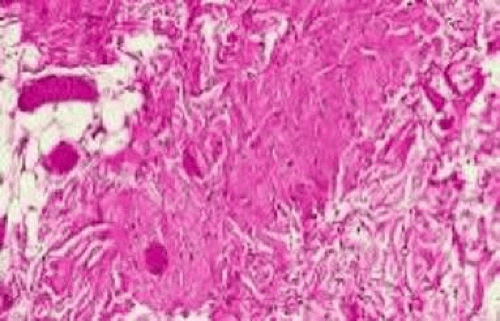 Figure 1. Histopathology of the levator palpebrae superioris of a patient with congenital ptosis demonstrates fibrovascular and fibroadipose infiltration. |
 Figure 2. Patient with congenital ptosis. Note the lack of eyelid crease. |
Concomitant maldevelopment of the superior rectus muscle seen in 5% of congenital myogenic ptosis cases is called double elevator palsy or monocular elevation deficiency. In these cases fibrofatty tissue deposition is seen in both muscles on biopsy.2,3 In these patients, congenital myogenic ptosis is associated with poor Bell phenomenon and/or vertical strabismus. Complete repair of congenital myogenic ptosis is impossible, because the pathophysiology involves abnormal muscle physiology. Therefore, surgical options for these patients involve bypassing the LPS muscle and utilizing the nearby frontalis muscle. These operations will be described in a subsequent section of this chapter.
Congenital Aponeurotic Ptosis
Rarely, congenital ptosis may be aponeurotic in origin. In this situation, the ptosis is caused by failure of the levator aponeurosis to insert at its normal position on the anterior tarsus. This may result from birth trauma, especially with forceps delivery. Usually there is good eyelid excursion, but an abnormally high eyelid crease is present, similar to adult-onset involutional or aponeurotic ptosis.
Congenital Mechanical Ptosis
Mechanical ptosis can result from a congenital abnormality causing mass effect in the upper eyelid, brow, or orbit. Congenital hemangiomas, dermoids, or plexiform neurofibromas are common causes of mechanical ptosis. In addition, amblyopia can result from astigmatism induced by the mass effect on the globe or from obstruction of the visual axis resulting from the ptotic eyelid. Hemangiomas usually resolve spontaneously or respond to steroid injections.2 Propranolol has been used successfully to treat pediatric hemangiomas.4;emoval of an offending dermoid will usually eliminate the associated mechanical ptosis. Ptosis associated with neurofibromatosis and plexiform neurofibromas is not easily corrected. Treatment often involves repeat debulking procedures.
Congenital Neurogenic Ptosis
Congenital neurogenic ptosis is rare and is caused by innervational defects during embryonic development. Marcus Gunn jaw-winking ptosis (MGJWP) is the most common type of congenital neurogenic ptosis. It is observed in 4% to 6% of patients with congenital ptosis. In MGJWP, abnormal innervation of a normal LPS muscle causes LPS co-contraction with jaw movement (Fig. 3). This ptosis and synkinetic movement results from aberrant connections between the motor branches of the fifth cranial nerve that normally supply the pterygoid muscle or the muscles of mastication and the superior division of the oculomotor nerve (CN III) that innervates the LPS and superior rectus muscles.5 The aberrant connections in the central nervous system can be bilateral, although typically only one side is symptomatic.6 Clinically, these patients usually have a baseline ptosis on the affected side in primary gaze from decreased relative baseline tone from the superior division of CN III. The wink is induced by use of muscles of mastication (e.g., sucking, swallowing, chewing, or a lateral motion of the jaw usually toward the contralateral side). The wink consists of a rapid elevation of the ptotic lid to a position equal or higher than the unaffected side and then an equally rapid descent back to the ptotic state.7 In this situation, ipsilateral hypotropia or vertical strabismus is common because the superior rectus muscle may be involved. Some children with the associated jaw wink develop adaptive behavioral mechanisms and learn to avoid movements that induce the wink reflex. This has caused some clinicians to state that MGJWP ptosis improves with age. If the jaw wink is clinically insignificant, it may be ignored in treatment of the ptosis. However, the jaw wink may be more exaggerated following ptosis correction. Jaw winking becomes more clinically significant when it is 2 mm or more; these cases of synkinesis can be addressed at the time of ptosis repair. Any attempt to repair the ptosis without addressing the jaw wink would result in an exaggeration of the abnormal eyelid movement to a level above the superior corneal limbus. Transection of the levator aponeurosis in combination with a frontalis suspension procedure will help to alleviate synkinetic eyelid movements after the ptosis repair.
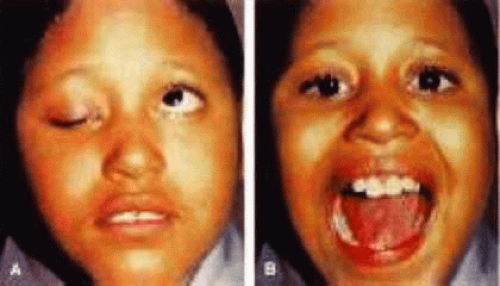 Figure 3. Marcus-Gun jaw-winking ptosis (synkinesis linking fifth cranial nerve to third cranial nerve ptosis). A. Baseline congenital ptosis of the right eye. B. On jaw opening, the right lid is elevated. (Adapted with permission from American Academy of Ophthalmology. Orbit, Eyelids and Lacrimal System; BCSC Section 7. San Francisco, CA: American Academy of Ophthalmology, 2000–2001: Figure XII-15.) |
Congenital third nerve palsy is another type of neurogenic congenital ptosis. In these patients, blepharoptosis manifests along with deficits in adduction, elevation, and depression of the globe resulting from the abnormal innervation by the oculomotor nerve to the LPS muscle, inferior rectus, superior rectus, medial rectus, and inferior oblique. Mydriasis may be present depending on the degree of decreased innervation to the pupillary sphincter muscle.
Congenital Horner syndrome is a rare type of neurogenic ptosis, and patients exhibit mild ptosis, miosis, anhidrosis, and decreased pigmentation of the iris and areola on the affected side.8 The ptosis is a result of decreased function of the sympathetically innervated Müller muscle of the upper eyelid. Decreased sympathetic tone to the inferior tarsal muscle results in elevation of the lower eyelid, further narrowing the palpebral fissure on the involved side, the so-called reverse ptosis of the lower eyelid. Although the lesion may be anywhere on the sympathetic chain, congenital Horner is most often the result of a second-order neuron involvement from brachial plexus injury at birth. The classification of the first-order (central), second-order (preganglionic), and third-order (postganglionic) neuron lesions along the sympathetic chain are discussed later in the chapter.9
Congenital Eyelid Syndrome
Another type of congenital ptosis that is inherited is the congenital eyelid syndrome, or the blepharophimosis, ptosis, epicanthus inversus syndrome (BPES). The inheritance is autosomal dominant and affects about 5% of patients with congenital ptosis3 (Fig. 4).
 Figure 4. Patient with blepharocanthal syndrome. Note the presence of ptosis, horizontal shortening of the palpebral fissures, and epicanthus inversus. |
Blepharophimosis—vertical and horizontal shortening of the palpebral fissure
Ptosis with poor levator muscle function and absence of a definitive eyelid crease
Epicanthus inversus—an epicanthal fold more prominent on the lower eyelid that extends to the upper eyelid
Telecanthus—a widened intercanthal distance
Lower eyelid ectropion
Associated findings with BPES include hypertelorism, low projection of the nasal bridge, hypoplasia of the superior orbital rims and the tarsal plate, and lop ears (deformity of the helical cartilage causing overhanging of the upper third of the ear).2 In addition, because hypogonadism and specific hormonal deficiencies may also occur, females with this condition should be closely monitored for menstrual irregularity and infertility.10 In cases of ptosis associated with coexisting congenital eyelid anomalies such as in blepharophimosis, eyelid reconstruction may be staged with the ptosis repair usually being delayed until the other eyelid abnormalities are corrected. Ptosis correction at the same time as eyelid reconstruction may be necessary in the setting of ptosis-induced amblyopia that threatens normal visual development.
ACQUIRED PTOSIS
Acquired ptosis is a change from the patient’s baseline. Upper eyelid anatomy is crucial in understanding the pathogenesis of acquired ptosis. (See Chapter 72.) The levator complex of the upper eyelid includes the LPS muscle and aponeurosis. The Müller muscle is a sympathetically innervated muscle consisting of smooth muscle fibers. It originates on the undersurface of the levator and inserts 15 mm inferiorly along the superior edge of the tarsus. The levator aponeurosis is composed of a group of collagenous lamellae that have attachments to the dermis and the anterior tarsal surface. These lamellae may become attenuated or dehisced from their attachment sites, resulting in an involutionally ptotic eyelid.11 Ptosis of the upper eyelid can also be caused by reduced elevating function of the eyelid retractors as a result of neurogenic, myogenic, mechanical, or traumatic insult.
Acquired Aponeurotic Ptosis
Acquired ptosis is most commonly aponeurotic in etiology. Involutional changes of both aging and repetitive traction on the eyelid result in levator aponeurosis thinning and disinsertion from its tarsal attachments. Although aponeurotic ptosis most often occurs in older adults, it can be seen in at any age. Younger patients may develop aponeurotic ptosis as a result of trauma, orbital or eyelid swelling, excessive eyelid rubbing, contact lens wear, pregnancy, blepharochalasis, prior ocular surgery, or chronic ocular inflammation.12 In these patients, the eyelid crease is usually elevated because of disinsertion from the tarsal plate with superior retraction of the levator aponeurosis (Fig. 5). The attached dermis and orbicularis move cephalad with the migration of the aponeurosis and lead to the formation or appearance of a superior sulcus deformity, a high eyelid crease and fold, and thinning of the upper eyelid.
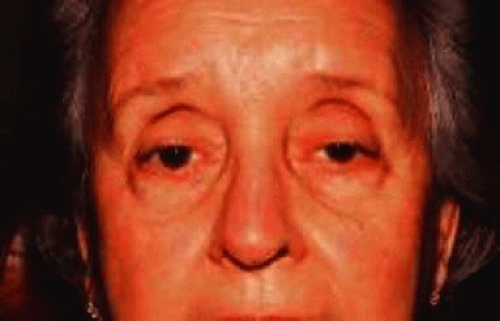 Figure 5. Patient with levator aponeurosis dehiscence. Note the raised eyelid creases bilaterally. |
Patients with aponeurotic ptosis often complain of eyelid and brow fatigue. The ptosis is usually worse later in the day owing to frontalis muscle fatigue, and this finding should not be confused with those of myopathic ptosis (i.e., myasthenia gravis [MG]). Patients often notice a decrease in their superior visual field as a result of the upper eyelid descent. Older patients often complain of difficulty reading because the ptosis is worse on downgaze, and again this is often worse at the end of the day. Involutional changes of the eyelids associated with aging include loss of tone of the LPS and Müller muscles, and atrophy of the orbital fat, leading to a mild degree of enophthalmos.3
The LPS muscle function in aponeurotic ptosis is either normal or slightly reduced, as assessed by measuring in millimeters the amount of eyelid excursion from downgaze to upgaze.7 These measurements are helpful in distinguishing congenital from acquired ptosis (Table 1). LPS function in normal individuals measures 16 to 20 mm. This is reduced to 12 to 16 mm in acquired aponeurotic ptosis. Horizontal laxity in the upper eyelid is also associated with aponeurotic ptosis and can be attributed to weakness in the lateral canthal tendon or the supporting eyelid structures of the upper eyelid tarsal sling. Specifically, dehiscence of the medial aspect of the Whitnall ligament can occur, resulting in loss of medial support of the eyelid. The tarsal plate shifts laterally, causing a more ptotic eyelid nasally and an abnormal eyelid contour. The lateral shift is usually associated with aging and should be recognized preoperatively, because this phenomenon may alter the surgical approach for ptosis repair. Lash ptosis can develop as a result of secondary ptosis from the hooding effect of redundant skin and soft tissue of the upper eyelid resting on the eyelashes. Lash ptosis may also be caused by aging changes in the upper eyelid, including decreased tone, loss of structural support from increasing laxity of the upper eyelid, disinsertion of levator aponeurosis attachments to the supraciliary dermis, and atrophy of the tarsus. Surgical repair in these patients with horizontal laxity is difficult and demands balance between horizontal tension of the eyelid and vertical pull of the levator aponeurosis. Correction of aponeurotic ptosis is accomplished most commonly by either an external approach levator resection or with an internal approach ptosis repair, both of which are discussed later in this chapter.
Table 1. Comparison of Congenital Myogenic Ptosis to Acquired Aponeurotic Ptosis | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
Acquired Neurogenic Ptosis
Acquired neurogenic ptosis is usually caused by a CN III (oculomotor nerve) palsy or by disruption of sympathetic innervation to the Müller muscle. There are two classifications of CN III palsies: vasculopathic (from diabetes, hypertension, or arteriosclerotic disease) and compressive (from a neoplasm or an aneurysm.) Vasculopathic processes rarely affect the pupillary fibers of the third nerve and generally resolve spontaneously within 3 months. It is critical to note that CN III palsies owing to an aneurysm may be evolving and may not yet involve the pupil. Certainly if a CN III palsy has not improved within 3 months or if the pupil is involved in the patient’s initial presentation, further workup to rule out neoplasm or aneurysm must be performed.2 A CN III palsy rarely causes an isolated LPS muscle deficit; frequently other extraocular muscles are involved. Pupillary fibers are more likely to be involved with a compressive neuropathy. The classic presentation of a compressive CN III palsy is unilateral, rapidly evolving ptosis, exotropia, hypotropia, and mydriasis. This presentation should immediately prompt further evaluation with an imaging study.
A patient with an oculomotor nerve palsy may develop synkinetic ptosis as a result of aberrant regeneration and misdirection of CN III fibers. Although more often congenital, this condition may also develop during recovery from an oculomotor nerve paralysis. Synkinesis can cause elevation of the eyelid with the use of the extraocular muscles, as well as paradoxical pupillary dilatation or constriction.9 The ptotic eyelid may rise as the inferior rectus, medial rectus, or superior rectus muscle contracts. If aberrant regeneration of the nerve is observed in the examination of a patient without a known history of oculomotor nerve palsy, a workup is necessary to rule out a compressive neoplasm of the cavernous sinus or an aneurysm.
Horner syndrome is caused by a disruption of sympathetic innervation to eyelid retractor muscles, to pupillary dilator muscles, and to the sweat glands in the skin on the affected side. Horner syndrome classically presents with unilateral ptosis, miosis, and anhidrosis. Paralysis of the Müller muscle in the upper eyelid results in mild ptosis measuring approximately 1 to 2 mm. The lower eyelid may be elevated approximately 1 mm because of paresis of the inferior tarsal muscle; this finding is also called reverse ptosis. This further narrows the palpebral fissure and may mislead the clinician to think that the patient manifests enophthalmos, or even contralateral proptosis. The pupil on the involved side is constricted and dilates poorly in the dark as compared with the pupil of the unaffected eye. There may be anhydrosis on the ipsilateral body in first-order, ipsilateral face in second-order (preganglionic), and ipsilateral suprabrow area in third-order (postganglionic) lesions.9 Iris depigmentation leading to heterochromia occurs in congenital Horner syndrome.
Various lesions located along the course of the sympathetic chain cause Horner syndrome. Tumors, inflammatory processes, aneurysms, and injuries are the most frequent causes.3 The anatomic location of the lesion determines if it is situated along a first-order, second-order, or third-order neuron. The central or first-order neuron courses from the hypothalamus to the cervical portion of the spinal cord.13 Lesions in the brain, spinal cord, or brainstem may result in a first-order insult. The second-order, or preganglionic, neuron passes from the cervical portion of the spinal cord along the sympathetic chain through the upper thorax and neck to synapse in the superior cervical ganglion. Apical lung tumors (Pancoast tumors), metastasis, thoracic surgery, thoracic aortic aneurysms, or trauma to the brachial plexus are examples of second-order lesions that can cause disruption of the sympathetic chain. The third-order, or postganglionic, neuron travels with the sympathetic fibers along the internal carotid artery to the orbit. These lesions are most often of a vascular cause, such as a carotid artery dissection. Third-order lesions also result from head and neck trauma, tumors, infections, and lesions of the cavernous sinus.14 Localization of the lesion along the sympathetic chain is essential for the proper diagnosis and treatment of Horner syndrome.
Topical pharmacologic testing can be used to clinically localize the lesion causing the Horner syndrome. Topical cocaine 4% to 10% is used to confirm the presence of Horner syndrome. Cocaine acts to block the reuptake of norepinephrine at the neuromuscular junction, thereby indirectly stimulating the pupillary dilator muscle. This blockade results in pupillary dilation in the normal eye. Cocaine similarly activates the Müller muscle, elevating the ptotic eyelid 1 to 2 mm. The typical response in a patient with Horner syndrome after the application of cocaine is a lack of pupillary dilation and failure of eyelid ptosis to resolve. However, addition of epinephrine or hydroxyamphetamine topically can dilate the pupil and elevate the ptotic eyelid in a third-order lesion. Since cocaine solution may be difficult to obtain, apraclonidine 0.5% or 1% (Iopidine) is a practical alternative to cocaine.15;harmacologic testing with apraclonidine is based on the law of denervation supersensitivity in which an organ that has lost its normal innervation will become more sensitive to its chemical neurotransmitter once exposed. Apraclonidine is a direct-acting alpha-1 and alpha-2 receptor agonist with little or no effect on the normal pupil. However, because of denervation supersensitivity and up-regulation of alpha-1 receptors, the patient with Horner syndrome will have dilation of the affected pupil in response to apraclonidine.
Topical hydroxyamphetamine 1% (Paredrine), is used to differentiate first and second order, or preganglionic lesions from a third-order or postganglionic lesion.16 The Paredrine test should be done more than 24 to 48 hours after a cocaine test to avoid inaccuracies. In a normal eye and in a preganglionic Horner syndrome, the postganglionic neuron is intact and norepinephrine is present. The action of topical hydroxyamphetamine releases the norepinephrine from the presynaptic terminal into the myoneural junction of the pupillary dilator muscle fibers, resulting in dilation of the pupil. In addition, hydroxyamphetamine acts similarly to stimulate the Müller muscle, which will raise the ptotic upper eyelid. A poor pupillary response or no improvement of ptosis usually indicates a third-order lesion, in which the postsynaptic terminal has degenerated and no norepinephrine is present. Hydroxyamphetamine has become increasingly difficult to obtain in the United States. Phenylephrine 1% causes a similar effect to that of hydroxyamphetamine 1% and is a useful alternative to the Paredrine test.17;henylephrine 1% causes dilation of the pupil in a patient with a third-order or postganglionic Horner syndrome but not the normal or preganglionic Horner pupil.
Horner syndrome is often associated with other neurologic signs, especially when caused by tumor, aneurysm, or inflammation. The etiology must be identified before initiating proper treatment. Surgery to correct eyelid ptosis is considered only when a thorough evaluation, proper diagnosis, and treatment has been implemented.
Myasthenia gravis (MG) is a B-cell mediated autoimmune disease that can cause a neurogenic acquired ptosis. MG is characterized by weakness of the affected muscles. Patients often initially present to an ophthalmologist with complaints of diplopia, ptosis or both, because the extraocular muscles, particularly the LPS, are often the first to be affected in new-onset MG.3 In MG, antibodies to acetylcholine (ACH) receptors cause a functional blockade of the ACH receptors at the neuromuscular junction. The histopathology in MG also demonstrates a flattening and marked loss of the folds present in the postsynaptic membrane. In the normal membrane, these folds increase the surface area available for ACH-receptor binding.18 The reduced number and availability of receptors causes a decrease in the total binding of ACH in the synapses, reducing neural transmission and resulting in a weakness of the levator muscle and ptosis. The unbound ACH in the synaptic cleft is then degraded. Ptosis may be exacerbated by prolonged upgaze, which causes saturation of the few remaining ACH receptors.
MG often initially presents as a neurogenic ptosis and must always be considered in the differential diagnosis of a patient presenting with an acquired ptosis. The eyelid ptosis often begins unilaterally but may variably affect the other eyelid. Other accompanying findings may include dysphagia, hoarseness, dysarthria, dyspnea, easy fatigability, as well as weakness of the jaw, neck, trunk, and arms.13 The dysphagia and dyspnea can be life threatening. Patients often note progressive worsening of symptoms resulting from fatigue as the day progresses.3 Symptoms are exacerbated after exertion and improve with rest. Patients diagnosed with MG occasionally have associated autoimmune disease. Five percent of patients with MG also have Graves disease. Many patients with MG have been found to have hyperactivity of the thymus gland. Thymic hyperplasia is thought to play a role in the immune mechanism in MG.
Ocular myasthenia is myasthenia isolated to the periocular muscles, including the LPS, the orbicularis oculi, and the other extraocular muscles. The symptoms are usually localized and include ptosis, diplopia, and fatigue of the periocular muscles. In this type of myasthenia, the ACH-receptor antibody normally found in myasthenic patients is often absent. This difference may be a result of the antigenic differences between the synaptic terminals of ocular muscles and other skeletal muscles. The extraocular muscles differ from other skeletal muscles in that they have multiple end plates for each muscle fiber and a myoneural junction that occurs within (hypolemmal) instead of on the muscle fiber. Ocular myasthenia may occasionally be seen with other autoimmune diseases, such as Hashimoto disease, hyperthyroidism, multiple sclerosis, systemic lupus erythematosus, polymyositis, or hemolytic anemia.19
The classic diagnostic test for myasthenic ptosis is the Tensilon test. Edrophonium chloride (Tensilon, Enlon) is an antiacetylcholinesterase drug that is administered intravenously. Pharmacologically, it acts to overcome the antibody receptor blockade in MG by inhibiting the breakdown of ACH. A positive test in a patient with myasthenia resolves the weakness of the affected muscle (e.g., LPS or rectus muscle) and results in resolution of symptoms (e.g., ptosis or diplopia). Patients must be forewarned of the potential adverse effects of the Tensilon test, which can include fasciculations, nausea, lacrimation, salivation, flushing, abdominal cramping, bradycardia, and even respiratory arrest. Atropine sulfate (0.4–0.6 mg) is a muscarinic antagonist and must be available to administer in case of the adverse reactions of edrophonium. Atropine sulfate blocks the action of ACH by binding to postsynaptic muscarinic receptors. Some physicians treat with atropine prophylactically by injecting the patient with 0.6 mg intramuscular (IM) before administering the edrophonium.
The Tensilon test can be administered in an office setting, provided that the clinician and ancillary help are comfortable and equipped to handle potential adverse reactions. Safer environments include the operating room (OR), ambulatory surgery center, or emergency room where resuscitation can be easily performed. Whenever a Tensilon test is planned, the patient should be supine, and resuscitation equipment should be readily available. The patient’s vital signs, including blood pressure and pulse, should be monitored throughout the procedure. Edrophonium is initially administered as a test dose, 2 mg (0.2 mL) and the full dose is slowly injected intravenously. The patient is observed for 60 seconds for elevation of the ptotic eyelid or improvement of extraocular movements. If there is improvement of symptoms after 1 minute, it is a positive test, and, thus, the test is terminated. If no response occurs, the remaining 8 mg is slowly administered and the patient is observed for 1 to 5 minutes. This dose can be divided into two 4-mg doses, which may reduce adverse effects.13 Unfortunately, edrophonium is becoming more difficult to obtain in the United States.
Neostigmine methylsulfate (Prostigmin) is another anticholinesterase drug that can be used as a pharmacologic diagnostic test for MG. Although the adverse reaction profile is similar to edrophonium, the common side effects are less severe (e.g., salivation, fasciculations, and gastrointestinal discomfort). This is useful in children and adults who may require a longer observation period than with the Tensilon test. Neostigmine is administered intramuscularly and is concurrently injected with atropine. The dosage formula is as follows: (weight [kg] × adult dose) divided by 70. The adult dose typically measures 1.5 mg with 0.4 mg atropine. Resolution of ptosis or diplopia in a positive test is expected to occur over a 30- to 45-minute time period.
Other nonpharmacologic tests for MG include the rest test and the ice test. The rest, or sleep, test is a clinical diagnostic test.20 This involves taking precise measurements of the patient’s degree of ptosis on presentation, then allowing the patient to rest quietly with eyelids closed for up to 30 minutes. Immediately after the patient is aroused, the ptosis measurements are repeated. Improvement of the ptosis with rest is highly suggestive of MG, because the receptors not occupied by antibodies are free to bind newly released ACH.13 The ice test is a safe diagnostic clinical test for MG that is easily performed in the office. An ice pack is placed over the affected ptotic eye for 2 minutes, and the patient is evaluated for resolution of ptosis. Myasthenic weakness is known to improve with lowering of the temperature of skeletal muscle. Cold is thought to affect the neuromuscular junction by decreasing cholinesterase activity and by promoting efficiency of ACH by eliciting depolarizations at the end plate.21 Kubis et al20 demonstrated that in 90% of patients with MG ptosis, there is an improvement of their ptosis with the ice test. Recent studies concur that the ice test is more effective than the rest test alone in diagnosing the ptosis of MG.22
Another diagnostic test for MG is a serum assay for antiacetylcholine receptor antibodies (ACHR Ab test). Binding antibodies are detected in approximately 90% of patients with generalized MG and in 70% of patients with ocular MG.13 There have, however, been false-positive reports with this test in patients with thymoma without myasthenia, Lambert-Eaton myasthenic syndrome, small cell lung cancer, as well as one third of the population older than 70 years.23 If the patient has not yet been diagnosed with MG or if the ophthalmologist is the first physician to suspect MG, the patient should be further evaluated by his or her internist for systemic involvement. Referral to a neuro-ophthalmologist is usually appropriate for a patient suspected to have MG. Serologic screening tests for thyroid dysfunction and autoimmune diseases and radiographic studies looking for a possible thymoma should be performed.
Patients with MG should undergo appropriate treatment with anticholinesterase medication, such as pyridostigmine bromide (Mestinon) and neostigmine (Prostigmin), corticosteroids, or other immunosuppressant agents under supervision of their internist or neurologist. In patients with enlargement of the thymus or a thymoma, a thymectomy is usually indicated. Some patients, especially younger individuals, have achieved disease remission following thymectomy.3 The systemic manifestations of MG can be life threatening and must be managed before considering surgical treatment of blepharoptosis. Patients often demonstrate poor eyelid closure resulting from weakness of the involved orbicularis muscles, which may lead to problems at surgery. Patients must also be warned that the ptosis may fluctuate and correction of ptosis is difficult.
Acquired Myogenic Ptosis
Myogenic ptosis is a rare type of acquired of blepharoptosis and can be found in localized or diffuse muscular disease, such as the muscular dystrophies. The muscular dystrophies are distinguishable by their mode of inheritance and clinical features. Blepharoptosis is a clinical sign of certain muscular dystrophies, including myotonic dystrophy, chronic progressive external ophthalmoplegia (CPEO), and oculopharyngeal muscular dystrophy.
Myotonic dystrophy is an autosomal dominant (chromosome 19) multisystem disorder with an onset usually in the second decade of life. Histologically, myotonic dystrophy is characterized by degenerative changes of skeletal muscle including disruption of myofilaments and sarcoplasmic reticulum, focal accumulation of mitochondria, and eventually atrophy and fibrosis of muscle.24 The distal musculature is usually the first to be affected. Myotonia is often the initial symptom, which results from delayed relaxation of skeletal muscle after contraction. Symptoms are exacerbated with cold, excitement, and fatigue. Patients often develop characteristic myopathic facies, presenting with blepharoptosis, ectropion with midface ptosis, frontal balding, and wasting of the temporalis and masseter muscles. Ocular findings include the ptosis and blepharospasm, ophthalmoplegia (similar to CPEO), pigmentary retinopathy, “Christmas tree” cataracts, and miotic pupils.2 Associated systemic findings include cognitive reduction, insulin resistance, cardiac conduction abnormalities, and testicular and uterine atrophy. Electromyography (EMG) is a diagnostic test demonstrating myotonic discharges.
Chronic progressive external ophthalmoplegia (CPEO) is another muscular dystrophy with several variants. Most cases are maternally inherited resulting from mutations in mitochondrial DNA, although cases with autosomal dominant and autosomal recessive inheritance have also been identified. Inherited CPEO has been linked to specific families of Italian and Finnish descent. At least one form of CPEO, however, has been found to have occurred as a sporadic mutation. On muscle biopsy, histopathologic examination reveals ragged red fibers, which represent accumulations of abnormal mitochondria within the myofibers.24 EMG findings can also be abnormal. Because the disorder is mitochondrial in origin, it tends to affect tissues with high oxidative needs: cardiac muscle, skeletal muscle, and the central nervous system.25
CPEO usually presents in the second to fourth decade, often initially with bilateral progressive ptosis (Fig. 6A). In addition to LPS muscle involvement, patients also develop a weakness of other periocular muscles including the rectus muscles and the orbicularis oculi muscles (Figs. 6B–D). Despite limited ductions from weakness of the extraocular muscles, binocular vision is usually preserved. Various types of CPEO are associated with systemic deficits such as retinal pigment degeneration, hyperacusia, dysphagia, ataxia, and proximal muscle weakness. The retinal pigment changes differ from retinitis pigmentosa in that they are mainly at the posterior pole with a mottled appearance of the pigment epithelium. Visual changes are mild if any.24 There is also a pediatric variant of CPEO, known as Kearns-Sayre syndrome, which includes cardiac conduction abnormalities (a significant surgical risk), retinal pigment degeneration, and bilateral ptosis.25
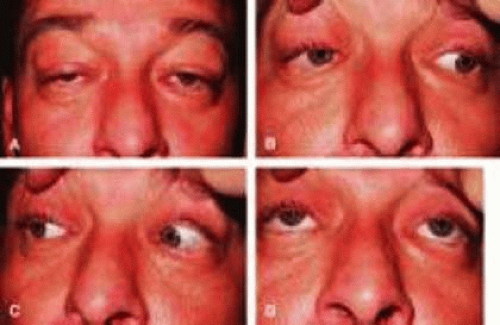 Figure 6. A. Patient with chronic progressive external ophthalmoplegia (CPEO) and ptosis. B–D. Extraocular movements are limited in fields of gaze.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|