Primary Bone Tumors of the Orbit
Dinesh Selva
Jack Rootman
Valerie A. White
Igal Leibovitch
Primary orbital bone tumors make up 0.6% to 2% of all tumors of the orbit.1,2,3 Their rarity, in conjunction with often similar histology in related lesions, has led to poor characterization of this group as a whole. Further, the microscopic and radiologic appearances occasionally differ from those seen in alternate sites. For the pathologist in particular, who frequently receives piecemeal or curetted specimens, the lesion needs to be viewed in the context of a clinical and radiologic background. Thus, close cooperation between clinician, radiologist, and histopathologist is required for an accurate diagnosis.
Our experience of more than 4,000 orbital cases seen at the University of British Columbia Orbital Clinic and the South Australian Institute of Ophthalmology (SAIO) during a 22-year period has yielded 98 cases of primary tumors of the orbital bones (Table 1). Two entities; fibrous dysplasia and osteoma, made up 41 of these cases and represent the conditions most likely to be encountered over a lifetime of clinical practice. In this chapter, we review the salient features of primary orbital bone tumors, adding our experience and suggesting a clinicopathologic classification.
Table 1. Clinicopathologic Classification of Primary Orbital Bone Disorders, UBC Orbital Clinic, 1976-1998 and South Australian Institute of Ophthalmology 1997-2007 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Clinical Patterns
Most primary orbital bone tumors belong to one of three categories.
Slow, Progressive, Noninfiltrative Mass Effect
The prototypical presentation in this group is that of a benign fibro-osseous lesion such as an osteoma. Proptosis, globe displacement, and deformity are the most common manifestations; the temporal course is measured in years. Any functional deficits, such as decreased vision or diplopia, are due purely to mechanical or compressive effects.
Subacute Mass Effect With or Without Sudden Soft Tissue Displacement
The reactive lesions of bone such as aneurysmal bone cyst, giant cell granuloma, and cholesterol granuloma often fall in this category. The evolution of the mass effect in these cases usually varies from weeks to months but can extend to years. In addition, they may be subject to intralesional hemorrhage, leading to sudden proptosis or displacement.
Relentless Progression of Mass Effect With or Without Infiltrative Features
Neoplasia, in the form of osteosarcoma, Ewing’s sarcoma, myeloma, and chondrosarcoma, frequently presents in this manner. The acuity of onset, with the exception of chondrosarcoma, is quicker than in the first category, and signs of infiltration, such as significant pain, decreased vision, or restriction of ocular movement, may be present.
Clinicopathologic Classification of Major Orbital Bone Tumors
The above clinical patterns correlate reasonably well with the following clinicopathologic classification (see Table 1).
Benign Fibro-Osseous and Cartilaginous Lesions
Osteoma
A true osteoma is a tumor-like mass of bony tissue that is histologically similar to normal bone. Its pathogenesis remains unclear, although traumatic, infective, or hamartomatous theories have been proposed.4 Others have suggested that osteomas arise exclusively at the junction of bones of cartilaginous and membranous origin.5 None of these theories account for the facts.
The most common sites of origin are the paranasal sinuses, skull, and facial bone. The estimated incidence of paranasal osteomas is between 0.014% and 0.43%; based on analysis of plain sinus radiographs.6 The true incidence may be higher, as the incidence on computed tomography
of the head or sinuses has been reported as high as 3%.6 In the sinuses, 50% occur in the frontal sinus, with the ethmoid, maxillary, and sphenoid involved in descending order of frequency. Most orbital osteomas are secondary invaders from adjacent sinuses, but on occasion they arise primarily in the orbit. In contrast to the sinus distribution, however, orbital osteomas appear to have a roughly equal origin from the ethmoid, frontoethmoid, or frontal regions.7,8 This may reflect the relatively thin barrier to expansion posed by the medial orbital wall.
Presentation
Most sinus osteomas are solitary and asymptomatic.6 However, when large enough to encroach on the orbits, a gradual evolution of proptosis or globe displacement over many years can occur (Fig. 1). There may be an associated headache as a result of expansion of the overlying cortex and periosteum, and a bony mass is often palpable in the superior or superomedial orbit. Obstruction of the sinus ostia may lead to chronic sinusitis or mucocele. Less common features include an acquired Brown syndrome,9 gaze-evoked amaurosis or pain,10,11 subluxation of the eye12 and erosion leading to orbital emphysema, or cerebrospinal fluid rhinorrhea.13 The sphenoid sinus, although a rare site, is significant because even a small lesion may lead to an orbital apex syndrome.
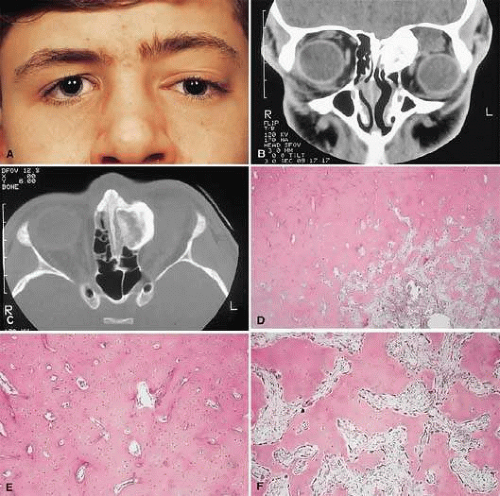 Figure 1. A. A 15-year-old boy with a 10-month history of left proptosis had an inferolaterally displaced globe and 6 mm of proptosis. B. CT scan showed a circumscribed sclerotic mass arising from the frontoethmoidal area, with a frontal mucocele laterally. C. Bone windows revealed the bony mass to have a cancellous core and a sclerotic periphery. D. The histology showed a peripheral zone of compact bone, with increasing osteoblastic activity and fibrous tissue toward the center (hematoxylin-eosin, 5×). E and F. Higher power revealed areas of compact bone with haversian canals similar to normal cortical bone and regions of trabecular bone (F) with more osteoblastic activity present (hematoxylin-eosin, 20×). |
An uncommon but important systemic association is Gardner’s syndrome. This autosomal dominant syndrome of osteomas, soft tissue tumors, and peripheral congenital retinal pigment epithelial hypertrophy also includes the development of colonic polyposis with subsequent malignant transformation.14,15,16 Multiple osteomas are common. Further, because bony lesions may predate the colonic pathology, patients with osteoma warrant a dilated funduscopy and referral to a gastroenterologist.17
Imaging
The plain radiograph and CT appearances consist of an osteoblastic round or ovoid sharply circumscribed mass, usually arising in the sinus and invading the orbit. Tumors growing in a sinus conform to its internal contour and often have a bosselated surface. Osteomas may be sessile or pedunculated and generally have a diameter of 1 to 5 cm.18 Bone window settings on CT imaging often show a very dense periphery with a more cancellous internal structure. However, the relative proportions of the two densities may vary with the size of the lesion.
Histopathology
It is important to distinguish osteomas from reactive osteomatous responses to infection, trauma, and chronic inflammation. The clinical and radiologic appearances are often invaluable in this regard.
Macroscopically, true osteomas have smooth or bosselated contours with a glistening white or pinkish coloration. A covering of mucoperiosteum or periorbita may be seen, depending on the site of origin.18
Osteomas have been classified histologically into three groups depending on the predominant tissue present: compact (cortical, ivory), cancellous (trabecular, spongy), and fibrous. Fu and Perzin19 have postulated that the histologic type is partly dependent on the age of the lesion, with the compact group representing the most mature and the fibrous the least. The fibrous subtype may, in fact, be part of a continuum incorporating ossifying fibroma and fibrous dysplasia.
The compact areas resemble normal cortical bone with dense bony areas and haversian systems. However, there are subtle differences in the arrangement of the haversian canals, which is often evident to the experienced bone pathologist. The cancellous areas consist of anastomosing trabeculae with an intervening fibrovascular stroma. Fatty and hematopoietic elements may also be present in the stroma, as well as evidence of osteoblastic activity along the trabeculae. The fibrous region is made up primarily of loose fibrovascular tissue with a few irregular bony trabeculae and osteoid elements.
In our series of surgically treated cases, we noted that although the three types of tissue were present in varying admixtures, in all cases there was a remarkably consistent pattern of arrangement. The most peripheral zone was made up of compact bone; moving toward the center or base of the lesion, there was an intermediate zone of increased osteoblastic activity, osteoid, and vascularity. The innermost region consisted of a loose fibrous stroma with a greater number of blood vessels, few trabeculae, and many plump osteoblasts. This configuration has been described previously by Albert and associates20 and illustrates the growth of these lesions.
The outermost zone presumably represents more mature bone, and the activity seen centrally suggests that this is where growth is initiated. This implies that extirpation of the central region is probably required to prevent recurrence. It may also explain why leaving residual peripheral areas do not usually lead to regrowth.
Finally, the histologic subtyping into compact, cancellous, and fibrous lesions probably has little practical significance, as there appears to be no correlation with the clinical course.
Management
Generally, asymptomatic osteomas can be treated conservatively. The only possible exception to this is in the sphenoid sinus; it is technically easier to remove a small lesion in this location before it has encroached on the orbital apex and optic canal.
If symptomatic and located in the anterior orbit, osteomas can be removed through an anterior orbitotomy. For anterior ethmoid tumors in the superomedial orbit, a modified Lynch incision is often used. Excision can sometimes be aided by coring the lesion and collapsing the cortex for removal. For more posterior tumors, involving the orbital roof or cribriform plate, a combined orbitocranial approach using a bicoronal incision has been favored, however, it carries a significant risk of morbidity.21
The endoscopic modified Lothrop procedure, with or without an orbital approach, has been recently described as an alternative for frontal osteoma excision. It involves the removal of the entire floor of both frontal sinuses, the upper nasal septum, and frontal sinus intersinus septum, allowing resection of the tumor. Stereotactic localization can be utilized to ensure preservation of the skull base. Although the endoscopic technique certainly entails a higher risk of leaving peripheral tumor than a frontal craniotomy, given that the chance of recurrence is rare even with a partial resection, so long as the central core is extirpated, it may be considered a viable alternative to avoid the morbidity of a craniotomy.
Fibrous Dysplasia
Fibrous dysplasia is a benign disorder in which proliferation of fibrous tissue and osteoid replaces and distorts medullary bone. The cause is unknown; past theories have included a maturation arrest at the woven bone stage, or hamartomatous proliferation.24 More recently, the discovery of a postzygotic mutation in the G protein in McCune-Albright syndrome suggests that the bone dysplasia in these patients is a manifestation of a somatic mosaic state.25,26
There are three forms of fibrous dysplasia: monostotic fibrous dysplasia (MFD), polyostotic fibrous dysplasia (PFD), and McCune-Albright syndrome.24,27 MFD accounts for 75% to 80% of cases, of which 20% affect the craniofacial bones. In the skull, the frontal bone is most commonly involved, followed by the sphenoid and ethmoid. Most patients with orbital involvement have MFD, although the disease has generally spread to contiguous bones by the time the patient comes to medical attention. PFD makes up 20% of all cases; half of these patients have head and neck involvement. McCune-Albright syndrome occurs largely in females and incorporates the triad of PFD, sexual precocity, and cutaneous pigmentation.28 This pigmentation appears as brown macules, usually six or fewer, with irregular “coast of Maine” borders.29
Fibrous dysplasia is generally recognized before age 30, although mild or asymptomatic cases may escape detection into late adult life.30 The gender distribution is roughly equal in MFD; there is a female predilection in PFD.
Presentation
The site and the extent of disease are the major determinants of symptomatology. Facial asymmetry, proptosis, and globe displacement evolving over many years are the most common manifestations (Fig. 2). Nasolacrimal duct blockage, diplopia, nasal obstruction, malocclusion, raised intracranial pressure, and cranial nerve palsies also occur.27,30,31,32 Acute or subacute compressive optic neuropathy can arise as a result of intralesional hemorrhage, sphenoidal mucocele, or secondary aneurysmal bone cyst.33 A more chronic visual loss, although less commonly reported, may occur as a result of compression in the optic canal or at the chiasm. On occasion, a superimposed ischemic neuropathy in the context of chronic compression leads to an acute on chronic deterioration in vision.34
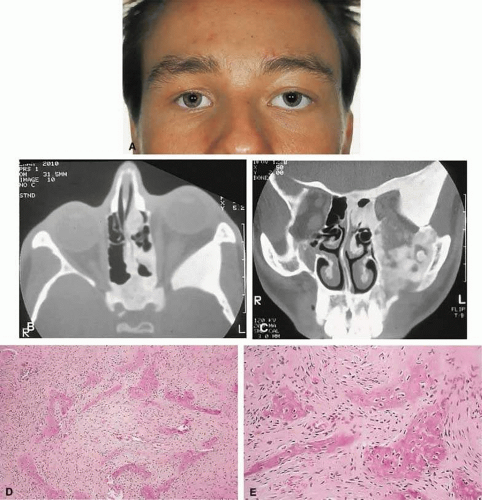 Figure 2. A. A 20-year-old man presented with a longstanding history of left proptosis and facial asymmetry. B and C. Bone window CT scan showed extensive fibrous dysplasia involving the greater wing of the sphenoid in a sclerotic fashion and a more pagetoid appearance in the maxillary and ethmoidal regions. D and E. Dominant histologic features consisted of irregular trabeculae of woven bone in a fibrous stroma with minimal osteoblastic activity (E) surrounding the osteoid (hematoxylin-eosin; D 20×, E 50×). |
Overall, the natural history is one of slow growth. Although this was previously thought to cease in adult life, there is evidence that fibrous dysplasia may progress well past the fourth decade.27
Rarely, malignant transformation to osteosarcoma, fibrosarcoma, chondrosarcoma, and giant cell sarcoma can occur; this is often signaled by a more rapid progression and increased pain. The incidence of this complication is estimated at 0.4% to 0.5%, rising to approximately 15% with prior radiation therapy.35
Imaging
In the craniofacial bones, fibrous dysplasia tends to expand the bone, with thinning of the overlying cortex. The margins are poorly defined, and the dysplasia transgresses suture lines; the proportion of mineralized to fibrous tissue determines the degree of radiolucency. Most cases demonstrate a relatively equal mixture, resulting in a pagetoid appearance. Where the fibrous element is predominant, there may be cystlike areas; a preponderance of mineralized tissue, however, results in a homogeneous, sclerotic, “ground-glass” picture. Fries36 reviewed 39 patients with fibrous dysplasia of the craniofacial bones and found a pagetoid pattern to be most common (56%), followed by sclerotic (23%) and cystlike (21%) appearances.
The primary differential is hyperostotic meningioma. This is distinguished by its occurrence in an older age group and by the presence of an associated enhancing soft tissue component, best seen on MRI. Also, meningioma often causes a more homogeneous thickening of bone, which in contrast to fibrous dysplasia does not leave a discernible cortical rim. MRI shows meningioma to have a signal isointense to gray matter on both T1- and T2-weighted images. Fibrous dysplasia, in contrast, tends to have a lower intensity on T1- and a heterogeneous signal on T2-weighted images.30,37 MRI may also have a role in fibrous dysplasia in the evaluation of mucoceles and recent hemorrhage.
On occasion, Paget’s disease and less commonly cystic bone lesions, such as localized Langerhans cell histiocytosis (eosinophilic granuloma), also enter into the differential diagnosis. Paget’s disease arises beyond the age of 40, is usually bilateral, and radiologically may show areas of cotton wool type density that are not usually seen in fibrous dysplasia.
Histopathology
Macroscopically, fibrous dysplasia consists of gritty, white-to-pink tissue, often with blood or serous-filled cystic areas. Histologically, there is a fibrous background containing trabeculae of woven bone. The stroma has variable amounts of collagen, fibroblasts, and vascularity. There may also be myxomatous areas and secondary aneurysmal bone cysts. The curvilinear bone trabeculae take on a variety of configurations, including C or Y shapes (so-called Chinese characters). These trabeculae sometimes have irregular margins as a result of the attachment of collagen fibers arising in the stroma. Cartilaginous nodules as well as small foci of lamellar bone are occasionally seen, but the vast majority of lesions contain immature woven bone. At its periphery, fibrous dysplasia permeates normal bone, and there may be areas of reactive bone with more prominent lamellar bone formation and osteoblastic rimming. Sequential biopsies of fibrous dysplasia from childhood to adult life have shown that the histologic picture does not change with time.38
In the skull, the major histologic differential is ossifying fibroma. The latter, however, is a more circumscribed lesion that displays prominent production of lamellar bone with osteoblastic rimming.
Management
Traditionally, there has been a conservative approach to surgery for fibrous dysplasia, with intervention reserved for gross deformity, functional deficits, pain, or sarcomatous transformation. The procedures included resection if the lesion was well localized, curettage with bone grafting, or contouring. The last two decades have seen a shift to more aggressive and earlier intervention. A multidisciplinary craniofacial approach has been advocated, wherein as much affected bone as possible is removed and the resulting defects are reconstructed in a single operation.39 The indications for intervention include those previously mentioned, with the added rationale of attempting to prevent complications such as optic nerve compression.25,40 However, long-term follow-up data comparing outcomes with the natural history of the disease are lacking. In addition, there have been two reports of blindness complicating prophylactic optic nerve decompression.27,34,41 A study of 38 patients with fibrous dysplasia of the lesser wing of the sphenoid bone, published in the New England Journal of Medicine, showed that encasement of the optic canal in fibrous dysplasia causes narrowing of the canal, but that in itself does not result in visual loss.42 The authors concluded that prophylactic decompression of the optic nerve does not appear to be indicated on the basis of the presence of fibrous dysplasia on diagnostic images alone, since it does not correlate with visual loss. Thus, the need for prophylactic treatment remains unresolved; but it is probably not recommended unless a functional deficit develops.
Ossifying Fibroma
There is controversy as to whether ossifying fibroma is a distinct clinicopathologic entity: some authors believe it is a variant of fibrous dysplasia. Nevertheless, there appear to be enough disparate features to characterize it as a benign fibro-osseous neoplasm.
Ossifying fibroma occurs most commonly in the mandible in the first two decades of life, with a proclivity for females. Only rarely does it arise in the orbit, with the frontal bone being most commonly involved, followed by the ethmoid and the maxillary bones. Review of the orbital cases reported in the literature shows an age range of several month to the fifth or sixth decades of life, and an approximately equal male/female ratio.12,19,43,44,45,46,47,48,49,50,51,52,53,54
Presentation
As a result of its slow growth, ossifying fibroma generally manifests as a gradual painless globe displacement, with a temporal course measured in years. The mass effect may also lead to proptosis, diplopia, and, if situated more posteriorly, compression of apical structures.
Imaging
Ossifying fibroma starts as a monostotic lesion that expands the bone of origin in a well-circumscribed manner. However, with growth it may spread to involve adjacent bones and may even extend across the midline to involve both orbits. The characteristic CT appearance is of a round or ovoid mass with a well-defined, thin sclerotic margin (Fig. 3). Centrally, there is often a patchy pattern of osteoblastic and osteolytic areas.48
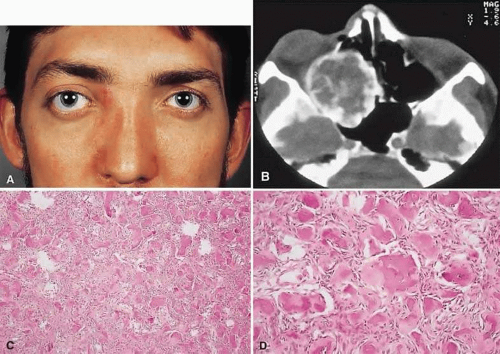 Figure 3. A. A 24-year-old man with a 10-year history of increasing right proptosis and a history of ossifying fibroma excised from the right ethmoid and sphenoid 12 years previously. Examination revealed 3 mm of proptosis and 2 mm of lateral globe displacement. B. CT showed a heterogeneous mass with a sclerotic margin involving the right ethmoid and orbit. C and D. After excision, the histology revealed a fibrous stroma containing small spherical ossicles characteristic of the psammomatoid variant of ossifying fibroma (hematoxylin-eosin; C 20×, D 50×). There has been no recurrence in 18 years of follow-up. |
Histopathology
Macroscopically, the lesional tissue is white to red and has a largely soft fibrous texture with variable grittiness, dependent on the amount of osteoid. Microscopically, it consists of a cellular vascular stroma containing trabeculae of lamellar bone. These bony trabeculae often have a thin surrounding of osteoid and, in contrast to fibrous dysplasia, display prominent osteoblastic rimming. There may also be osteoblasts as well as a few foci of giant cells in the stroma. If larger specimens are available, they may demonstrate a zonation phenomenon, seen as an increasing maturity of bone toward the periphery.19
In the psammomatoid variant described by Margo et al.,51 at least half of the tumor contains spherical ossicles. This histologic pattern has been correlated with a more aggressive local behavior and a tendency to recur after incomplete excision.
Management
The natural history of ossifying fibroma is one of inexorable progression; thus, surgical intervention is generally required. Because the propensity for recurrence after incomplete excision is well recognized, the surgical objective should be complete removal. This is particularly applicable in the psammomatoid variant. For anterior, relatively small lesions, this may be achieved using a percutaneous or bicoronal approach. However, most tumors tend to be sizable (5 cm in diameter) at presentation.51 Thus, for these lesions as well as those located more posteriorly, combined orbital, neurosurgical, and rhinologic approaches are usually necessary.47,53
Osteoblastoma
Osteoblastoma is a benign tumor composed of osteoblasts that produce osteoid and bone. It usually arises in the vertebrae and long bones, and its occurrence in the craniofacial region is extremely rare. These tumors are most commonly seen in the second and third decades and have a male/female ratio of 2:1.55
Less than 20 cases with orbital involvement have been reported in the past 40 years.19,56,57,58,59,60,61 In most cases the tumors appeared to arise from the orbital roof or ethmoid sinuses. The natural history is of slow growth, although some cases display a more aggressive behavior (aggressive osteoblastoma).
Presentation
The presentation is usually of a slowly progressive mass effect with proptosis and either downward or outward displacement of the globe. Pain or discomfort was a feature in several patients.
Imaging
In the long bones, osteoblastomas produce cortical expansion and have a lytic center. They can also simulate a large osteoid osteoma, with a lucent halo and central ossification. The different morphology of the orbital bones means that the tumor appears as an osteolytic lesion with a sclerotic margin; it occasionally has ossification of the matrix.55
Histopathology
The gross appearance is of a relatively gritty or friable, reddish-brown tissue. There is a broad spectrum of histologic appearances. The typical picture is of a network of osteoid trabeculae with osteoblastic rimming. These osteoblasts generally have abundant cytoplasm and regular nuclei. However, in some tumors, large epithelioid osteoblasts or a pseudosarcomatous appearance can be observed; this can lead to confusion with osteosarcoma.62 In contrast to osteosarcomas, however, even atypical osteoblastomas show a tendency toward peripheral maturation and do not permeate surrounding bone. Some authors have suggested that this atypical appearance may correlate with a more aggressive clinical course and have used the term aggressive osteoblastoma to define a separate clinicopathologic entity. It is a rare variant, with only very few cases being reported in the skull.63,64
The histology of osteoblastoma is similar to that of osteoid osteoma, with the latter being distinguished by a size smaller than 1.5 cm as well as a somewhat less cellular and vascular stroma.65 Nevertheless, they may represent a spectrum of disease, a fact somewhat supported by the finding of complex clonal structural changes in both tumors.66 Osteoid osteoma, however, is extremely rare in the orbit.67
Management
Excision is generally curative; however, there is one report of recurrence of an orbital tumor after a piecemeal removal.59 There have also been descriptions of a benign osteoblastoma of the skull that developed into an osteosarcoma after an incomplete excision,68 as well as a case of aggressive osteoblastoma of the temporal bone.69 In view of this, osteoblastomas should be completely removed under direct vision, where possible, to determine the margins. This usually entails an orbitocranial approach for tumors of the roof and a combined orbitorhinologic approach to those arising in the sinuses.
Chondroma
These benign cartilaginous tumors usually occur as asymptomatic lesions in the sinuses and nasal cavity. They rarely occur in the orbit, where they present as slow-growing, painless, firm lumps, often near the orbital rim or the trochlea.70,71,72 They have on occasion also been described in the soft tissues of the orbit.73 Radiologically, they are seen as well-circumscribed, dense masses that histologically consist of lobulated mature hyaline cartilage. Mature chondrocytes are seen in the cartilage, along with a variable fibrous or myxoid stroma. Surgical excision is always curative.70,74
A variety of other benign cartilaginous tumors, including osteochondromas, enchondromas (Fig. 4), and fibrochondromas, have also rarely been described in the orbit, although the histologic documentation is not always convincing.75
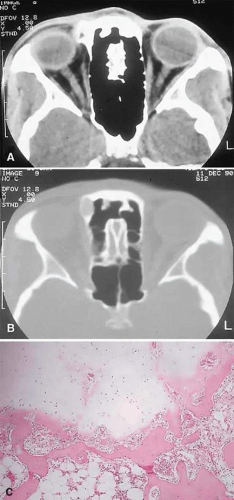 Figure 4. This 25-year-old woman had a 6-month history of a hard lump, which was readily palpable at the superomedial right orbital rim. A and B. CT showed a 1-cm bony mass that was more radiolucent centrally on bone window setting. C. The histopathology consisted of globules of mature cartilage encased in lamellar bone, leading to a diagnosis of enchondroma (hematoxylin-eosin, 20×). |
Reactive Lesions
Cholesterol Granuloma
A cholesterol granuloma is a foreign body response to the presence of crystallized cholesterol. The common sites are the middle ear and pneumatized portions of the temporal bone.76 In the orbit, it occurs almost exclusively in the diploë of the frontal bone overlying the lacrimal fossa, although it has also been reported in the zygoma.77
Theories of pathogenesis include a purely traumatic intradiploic hematoma or a hemorrhage occurring in a pre-existing bony anomaly. A breakdown of blood products then leads to cholesterol deposition and a granulomatous response. An analysis of reported cases of orbital cholesterol granulomas revealed a marked preponderance of men in the fourth and fifth decades of life.77,78,79,80
Presentation
A superolateral mass effect encompassing weeks to years is the typical mode of presentation. This leads to inferior globe displacement, proptosis, and diplopia in upgaze (Fig. 5). There may be associated headache or pain; one third of patients recall a prior trauma.77,78,79,80
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


