Chapter 67 Pharmacotherapy of Age-Related Macular Degeneration
Introduction
Age-related macular degeneration (AMD) is a spectrum of related diseases that have in common the progressive decline of vision as a consequence of dysfunction of the central retina and its underlying supporting elements, principally the retinal pigment epithelium (RPE) and choroid in older adults.1 Although the disease is seen in all racial and ethnic groups, it is more commonly encountered in females and light-skinned individuals and typically presents after the seventh decade.2–5 The disease has been traditionally classified into early and late stages.1
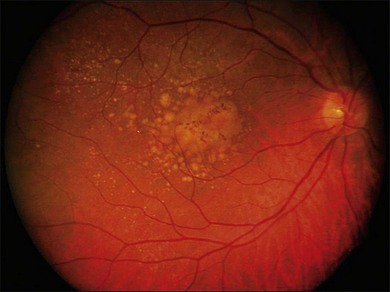
The early stages consist of alterations in the coloration of the macular pigment epithelium, both hypo- and hyperpigmentation, and the presence of drusen of greater than 125 µm in diameter. The early phases have been variously referred to as atrophic, nonexudative, preangiogenic, or dry AMD (Fig. 67.1). In contrast, exudative or neovascular AMD is typically a later-onset phenomenon, occurring in eyes with high-risk characteristics, including the presence of extensive soft drusen, Bruch’s membrane thickening, and focal hyperpigmentation. This phase of the disease is characteristically accompanied by rapid loss of vision over a period of 6–12 months and the development of a central disciform fibrotic scar. Without intervention, the visual acuity generally decreases to the range 20/200 or worse over the 12 months following the onset of this phase.3,5 In light of major improvements in therapy for this condition in the past 5–10 years, previously employed forms of treatment including thermal laser photocoagulation and surgery are now viewed to have limited benefit for this condition, particularly when it presents with involvement of the center of the fovea.6–10 In recent years, AMD therapy has become increasingly targeted toward specific molecular pathways. The discovery of multiple genes and markers for AMD and neovascularization has identified a significant number of new targets for pharmacotherapy.
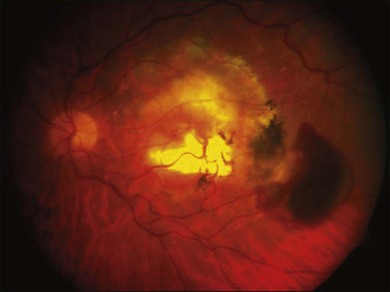
Relevant to the purpose of this chapter, it is important to understand that the mechanisms of vision loss in AMD differ according to the stage, and that different stages may be amenable to different points of attack.2,5 Importantly, at least two separate and distinct mechanisms for vision loss occur in exudative AMD: (1) proliferation of new vessels accompanied by secondary fibrosis and disorganization of the pigment epithelium and outer retina, which is a somewhat gradual process; (2) secondary alterations in the permeability of retinal and choroidal vessels accompanied by retinal pigment epithelial dysfunction leading to the accumulation of serous or serosanguineous fluid beneath the RPE, neurosensory retina, or within the retina itself, and are associated with more acute visual dysfunction. Preventing the vision loss associated with advanced forms of nonexudative disease has proven more challenging. In recent years an increased understanding of the mechanisms underlying drusen formation, pigment epithelial senescence, and loss of photoreceptors and choriocapillaris has made it more feasible to address therapeutically the visual dysfunction arising from the atrophic form of the disease (Fig. 67.2).
Etiologic factors
A variety of factors are thought to play a role in the development of AMD, including genetic susceptibility and a host of potentially modifiable environmental factors, including comorbidities, diet, medications, and light exposure.11
Genetic susceptibility
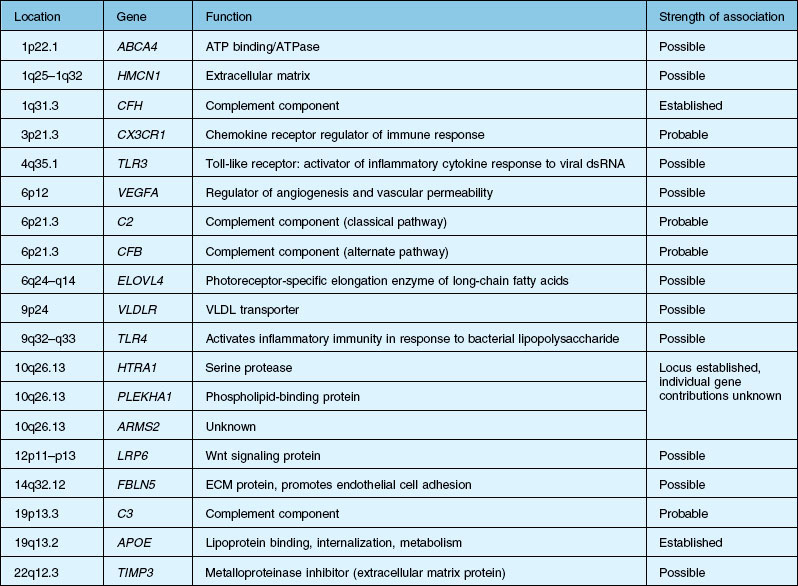
Over the past decade, high resolution genome scans for susceptibility loci in populations enriched for either choroidal neovascularization (CNV), geographic atrophy (GA), or both suggest susceptibility loci on numerous chromosomes. A number of candidate genes and their functions are listed in Table 67.1.2,12–15 An exhaustive discussion of the genetics and postulated mechanisms of ARMD is beyond the scope of this chapter; however, several genes deserve mention.
A significant breakthrough in the study of AMD genetics came in 2005, when it was discovered that single nucleotide polymorphisms (SNPs) in the regulation of the complement activation locus (RCA) of chromosome 1q31.3 increased the risk of AMD. The RCA locus contains the gene encoding complement factor H (CFH). The tyr402his protein polymorphism increased the risk of AMD between 2.7- and 7.4-fold and appeared to account for between 43% and 50% of the attributable risk of AMD. Additionally, it has been found that CFH genotypes may influence an individual’s response to certain treatment modalities and medications, including bevacizumab, photodynamic therapy with verteporfin, as well as AREDS (Age-Related Eye Disease Study) nutritional supplementation.16–20 CFH is responsible for downregulating activation of the complement system, including c5b-9, known as the membrane attack complex (MAC). MAC is thought to be important in the immune-mediated damage to Bruch’s membrane and an important pathogenic component of drusen.21,22
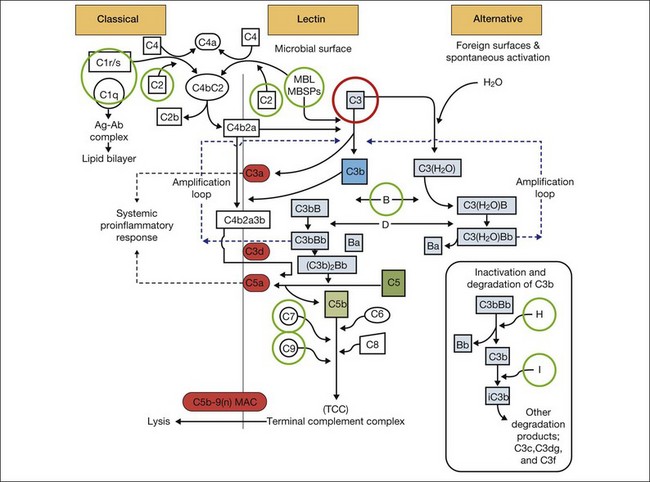
It is clear that both immune dysregulation and inflammation play key roles in the development and progression of AMD. Two recent studies found protective associations against the development of advanced AMD in those with genetic polymorphisms of the complement components C2 (OR 0.35–0.46) and complement factor B (CFB) (OR 0.35–0.44), while C3 (OR 2.66–3.9) was strongly associated with the development of GA.13,23 Figure 67.3 illustrates other complement components implicated in the pathogenesis of AMD. Additionally, elevated levels of the acute phase reactant CRP have been found to correlate with neovascular AMD. It is still unclear how genotypic variations in complement lead to an increased inflammatory state and the AMD phenotype. However, clinicopathologic specimens demonstrate the presence of inflammatory cells in Bruch’s membrane, and of bioactive fragments of C3 (C3a) and C5 (C5a) in drusen of AMD eyes and elevated VEGF expression in RPE cells.24,25 The inflammatory cascade is further characterized by retinal microglial activation and choroidal macrophage infiltration.26,27
The gene products for tissue inhibitor for metalloproteinase 3 (TIMP-3, chromosome 22) and EFEMP1 (chromosome 2, encoding an elastin-related protein, fibulin 3), which act to inhibit the stimulatory effects of VEGF on vascular cells, have been independently associated with advanced AMD in genome scans mentioned previously.28,29 Multiple distinct alleles in exon 5 of the gene encoding TIMP-3 have been causally linked to an autosomal dominant form of exudative macular degeneration with strong phenotypic similarities to exudative AMD,30–32 although it is not thought to account for the majority of patients with exudative AMD.33 TIMP-3 is also found in increasing quantities and is associated with thickening of Bruch’s membrane during aging, including within drusen and the RPE.34–36 An Arg to Trp mutation in EFEMP1 is thought to cause the abnormal accumulation of extensive drusen in malattia leventinese, an inherited macular degenerative disease with phenotypic characteristics closely correlated with advanced nonexudative AMD.34,37 The increased frequency of genome alterations coding for these two proteins (TIMP-3 and fibulin), which are important in the regulation of the extracellular matrix, particularly Bruch’s membrane, provides support for the hypothesis proposed by Blumenkranz that generalized increased susceptibility of elastic fibers to photic degeneration is an important risk factor for choroidal neovascularization.3
Early onset macular retinal degenerations such as Stargardt disease, characterized by the accumulation of lipofuscin within the RPE, have shed light on both the pathophysiology of RPE apoptosis and potential therapeutic avenues.38,39 While initial reports also suggested that the ABCR gene linked to Stargardt disease might also be a dominant susceptibility locus for AMD, subsequent investigators have been unable to confirm this apparent increased frequency.40,41 Nonetheless, increased amounts of lipofuscin are present in both Stargardt disease and in the earlier stages of the nonexudative AMD, and both diseases are characterized by GA in their later stages, suggesting that an understanding of the common pathways expressed by these two different diseases may provide some pharmacologic insights. Biochemical studies on the lipofuscin found in both Stargardt disease and AMD confirm that the principal component is N-retinylidene-N-retinylethanolamine (A2E), which accumulates in cells of the RPE and results in RPE apoptosis, photoreceptor death, and vision loss.22,42,43 This process is thought to be initiated by light activation of rhodopsin in the course of normal visual transduction as the first reactant in A2E biosynthesis. Isotretinoin (Accutane, discussed later), which slows the synthesis of 11-cis retinaldehyde and regeneration of rhodopsin, is able to slow the accumulation of lipofuscin in ABCR knockout mice and also aged wild-type mice.42 This suggests that compounds with mechanisms of action similar to isotretinoin, which is structurally related to vitamin A, may represent another therapeutic avenue for the slowing or prevention of AMD.22,39,41–43
There is additional discussion of AMD genetic susceptibility in Chapter 64 (Pathogenetic mechanisms in age-related macular degeneration.)
Environmental factors
Diet
The principal environmental factors thought to be associated with age-related macular degeneration include diet, history of smoking, light exposure, and use of supplemental medications. Recent AREDS reports have established a relationship between dietary intake of omega-3 long-chain polyunsaturated fatty acids and reduced risk of neovascular AMD (OR 0.61), as well as progression from drusen to GA.44–46 A large case control study suggested that higher dietary intake of carotenoids, particularly lutein and zeaxanthin, was associated with a lower risk of AMD, with the highest quintile having a 43% reduction compared with those in the lowest quintile. In that study no reduction could be found with the use of oral vitamin A, vitamin E or vitamin C.47 Subsequent studies suggest that the use of foods, particularly fruits rich in antioxidants and carotenoids, when consumed at the level of three servings or more per day, result in a pooled multivariate reduced relative risk of 0.64 compared with persons who consumed less than 1.5 servings per day.2 However, there have been a number of studies that have demonstrated little to no benefit with dietary regimens.28,48–50,51
Smoking*
Most studies have consistently implicated smoking as a statistically significant risk factor for the development of late-stage AMD.2,54 In a meta-analysis of three pooled studies from the United States, The Netherlands, and Australia, smoking was one of only two significant associations identified with incident AMD along with total serum cholesterol. Current smoking is associated with a 4.55-fold increased risk of neovascular AMD (vs. “never” smokers) and a 2.54-fold increased risk of atrophic AMD (vs. “never” smokers).55 Based on a pooled analysis of data, the risks of both neovascular and atrophic AMD seem to decrease once one stops smoking.55
Light exposure
Although there is a considerable body of compelling experimental data to suggest that increased retinal irradiance is positively correlated with an increased likelihood of advanced AMD, the epidemiological evidence supporting this hypothesis is modest.3,56,57 In the Beaver Dam eye study, participants exposed to summer sun for more than five hours a day during selected periods of time were at increased risk for increased retinal pigment (relative risk 3.17) and early AMD (relative risk 2.14) after 10 years of follow-up compared with those exposed to less than 2 hours per day.57
The underlying rationale for the hypothesis that increased light stress predisposes to AMD rests on the role of oxidative stress inherent with photo bleaching of the photoreceptors and, to a lesser extent, pigment epithelium. In particular, reactive oxygen intermediates (ROI) including hydrogen peroxide, singlet oxygen, and other short-lived species, which arise as the byproducts of cellular metabolism, are known to have a toxic effect on cellular membranes.48 It is felt that the cumulative effects of chronic low levels of these species result in profound damage to the retina and pigment epithelium through lipid peroxidation, mitochondrial DNA damage and induction of apoptosis.2,48 Experimental studies have confirmed the deleterious effect of chronic light and in particular blue and ultraviolet light on the RPE in part through the creation of A2E oxiranes, a major component of lipofuscin. The presence of natural antioxidants derived from dietary sources including lutein, zeaxanthin, lycopene, and ascorbate are thought to mitigate the effects of photo-oxidation by quenching free radicals and other intermediate species.48 It has been hypothesized that this represents one, but not the only mechanism of action, of the antioxidants in the AREDS study.58
Use of medications
Meta-analyses of pooled prospective studies suggest that certain medications may be associated with an increased or decreased risk of age-related macular degeneration. Antihypertensive medications, particularly beta-blockers, are associated with modest increased risk, whereas hormone replacement therapy in women, and tricyclic antidepressants, confer some relative protection.59 These observations may be able to be further exploited in the development of new classes of drugs in addition to avoiding potentially harmful drug interactions.
The reader is referred to Chapter 63 (Epidemiology and risk factors for age-related macular degeneration).
Systemic risk factors
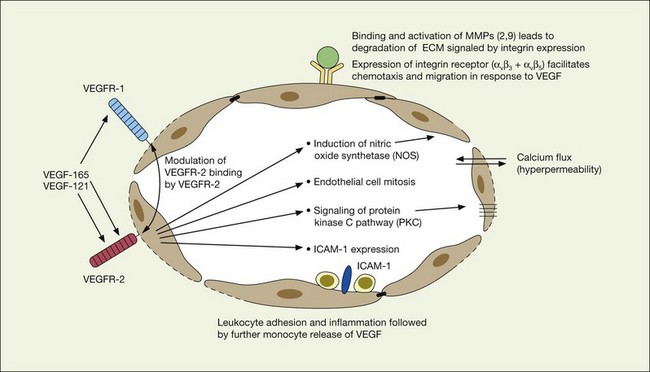
As previously noted, there appears to be a strong association between levels of ocular inflammation or systemic inflammatory disease and age-related macular degeneration. It has long been known that there is a relationship between VEGF and inflammation.2,60 Inflammatory foci and white cells are commonly seen in the choroid in autopsy specimens of patients with CNV.61–65 To the extent that inflammation does have a causative effect on the development of AMD, therapy aimed at reducing inflammation through complement inhibition or regulation, immunomodulation or stabilization of intracellular organelles such as lysosomes and resultant proteolysis, may be successful pharmacologic strategies. In animal models, intravitreal triamcinolone acetonide has been shown to inhibit preretinal and choroidal neovascularization induced by laser injury.66 There is increasing evidence of the linkage between VEGF, inflammation, and intracellular adhesion molecule 1 (ICAM-1) (Fig. 67.4). Triamcinolone modulates permeability and intracellular adhesion and ICAM-1 expression in cell culture, and it has further been shown that upregulation of ICAM-1 and invasion by CD18 positive leucocytes follows laser injury and precedes neovascularization.67,68 Comparable laser injury in ICAM-1 knockout mice demonstrated considerable reduction in neovascularization, confirming the likely participation of this class of molecules in VEGF-mediated angiogenesis. Other therapies that target CD18-positive leucocytes or ICAM-1 are likely to be beneficial in reducing the angiogenic stimulus associated with inflammation and aging.
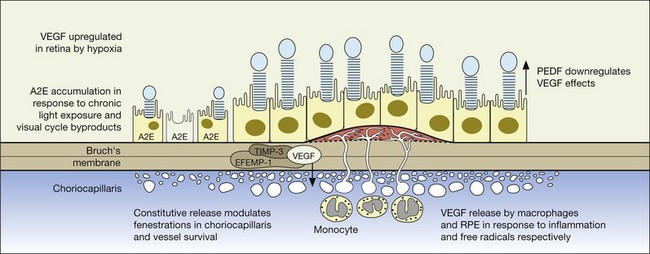
The pathophysiology of exudative amd: the crucial role of cytokines
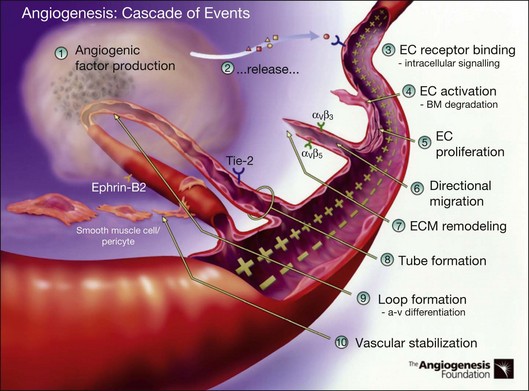
Definition and steps in angiogenesis
Angiogenesis refers to the creation of new blood vessels from existing blood vessels, and therefore is contrasted from the process of vasculogenesis seen characteristically in utero in which vessels are created de novo. The angiogenesis cascade has been characterized as occurring in multiple sequenced steps (adapted from The Angiogenesis Foundation, www.angio.org/understanding/process.php [current as of February 15, 2012]):
1. Release of angiogenic growth factors by diseased tissue.
2. Binding of the angiogenic growth factors to adjacent existing vascular endothelial cells (EC).
3. Activation of EC to produce molecules, including enzymes.
4. Dissolution of the surrounding basement membrane by activated enzymes.
5. Proliferation and migration of EC.
6. Adhesion molecules (integrins) help to serve as framework for advancing EC.
7. Further dissolution of tissue and remodeling with matrix metalloproteinases.
8. Formation of vascular tubes by EC.
9. Formation of blood vessel loops from EC tubes.
10. Stabilization of new blood vessels by smooth muscle cells and pericytes. Antiangiogenic therapies currently under investigation are thought to inhibit angiogenesis at various steps in this process (Fig. 67.5).
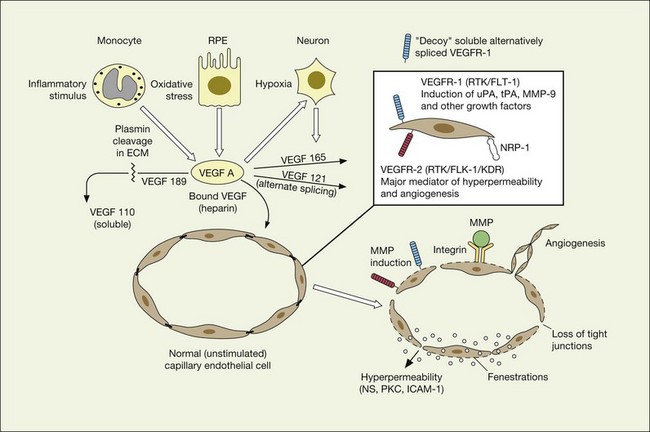
VEGF and other positive and negative modulators of angiogenesis
Regardless of the inciting stimulus involved in the development of pathologic neovascularization, it is now well established that VEGF plays a principal role, including not only the neovascular forms of age-related macular degeneration, but also diabetic retinopathy, iris neovascularization, and retinopathy of prematurity. Additionally, other cytokines may play an important role as well, including fibroblast growth factor (FGF), pigment epithelial-derived factor (PEDF), the integrins, angiopoietins, and matrix metalloproteinase inhibitors.2,30,69–72
VEGF undoubtedly represents the putative factor X first postulated by Michaelson associated with pathologic neovascularization.2 First described as a vasopermeability factor associated with tumors, the molecule has since been cloned and well characterized.73,74 VEGF has two important characteristics relevant to its role in the pathogenesis of the neovascular forms of age-related macular degeneration: (1) induction of angiogenesis through endothelial proliferation, migration, and new capillary formation, and (2) enhancement of vascular permeability.
Vascular permeability
VEGF produces important increases in hydraulic conductivity of isolated microvessels that are mediated by increased calcium influx and likely changes in levels of nitric oxide caused by induction of nitric oxide synthetase (NOS). While VEGF is both necessary and sufficient when administered exogenously to induce both these effects in vitro and in vivo, it is also well known that a number of other growth factors participate in this process. Some represent steps in a cascade initiated by VEGF while others act upstream.75,76 The chemical structure of VEGF is that of a heparin-binding homodimeric glycoprotein of 45 kDa.73 VEGF has significant homology to platelet-derived growth factor (PDGF). The human VEGF gene is organized into eight exons separated by seven introns and is localized in chromosome 6p21.3. Although there is only a single gene for VEGF-A, alternate post-translational exon splicing results in the generation of between four and six different isoforms having 121, 145, 165, 183, 189, and 206, amino acids respectively after signal sequence cleavage72,73 (Fig. 67.6). VEGF165 exists in both soluble and bound forms73 (Figs 67.6, 67.7), and is thought to be predominantly responsible for pathologic neovascularization.
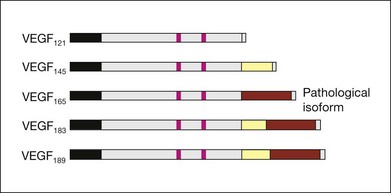
Although VEGF inhibitors share in common an attempt to mitigate the proliferative and permeability effects of VEGF on normal and neovascular tissue, there are likely to be differences in both efficacy and safety related to the choice of agents for several reasons. From a theoretical standpoint, since VEGF165 is the principal isoform involved in pathologic neovascularization, it has been argued that leaving the other three principal isoforms intact (121, which is principally diffusible in soluble form, and 189 and 206 which are principally matrix-bound through high affinity heparin binding sites), avoids the possibility of interference with normal homeostatic mechanisms associated with constitutive VEGF expression. Indeed, previous animal studies suggested that blockade of VEGF164 in the mouse (equivalent to VEGF165 in humans) is as effective as nonselective antibody-mediated VEGF blockade in preventing pathologic neovascularization in a hypoxia-induced angiogenesis model, while not interfering with normal physiologic development of retinal vasculature.60,77 However, it has become apparent with widespread use of bevacizumab and ranibizumab (monoclonal antibodies against multiple VEGF isoforms) that nonselective blockade has few untoward effects on normal retinal vascular physiology.
VEGF receptors
VEGF exerts its effects on cells through two highly related receptor tyrosine kinases (RTKs) VEGFR-1 and VEGFR-2.73 VEGFR-1 also termed FLT-1 (FMS-like tyrosine kinase) was described first, but its role remains open to debate. Like VEGF, VEGFR-1 is upregulated by a hypoxia-inducing factor (HIF)-dependent mechanism. The receptor undergoes weak tyrosine autophosphorylation in response to VEGF. It is thought not to be primarily a mitogenic stimulus but rather a “decoy” receptor, which downregulates the activity of VEGF by sequestering and rendering the factor less available to VEGFR-2 (Fig 67.7, 67.8). This factor may be most important during embryogenesis rather than during pathologic neovascularization as well as in hematopoietic bone marrow-derived cells and neural signaling.73,78
VEGFR-2 (KDR in humans or FLK-1 in mice)
A large body of evidence exists to support the critical and probably rate-limiting role of VEGF in neovascular forms of age-related macular degeneration. High messenger RNA levels and increased VEGF receptor levels are observed in areas of CNV in primates as well as in the extracted neovascular membranes of patients removed at surgery and following autopsy.2,63 Additionally there are strong indications that elevated levels of VEGF are the proximal cause for the hyperpermeability seen not only in diabetic macular edema, but in patients with subsensory and intraretinal fluid associated with CNV as well. The permeability changes resulting from VEGFR-2 are thought to be mediated to a large extent by endothelial nitric oxide synthetase-based generation of increased nitric oxide levels, and associated changes in calcium flux (Fig. 67.8). These alterations can be reversed either by direct blockade of VEGF, the receptor, or NOS using a knockout model.2,74,75 Inactivation of soluble VEGF by monoclonal antibodies directed against it, or inhibition of ICAM also appear to be effective as well. In addition to either the indirect inhibition of VEGF effects through modulation of ICAM, or indirect effects on nitric oxide phosphorylase, nitric oxide synthetase, or protein kinase C, another modulator of permeability, there are several other methods by which VEGF effects can be blocked in the eye and elsewhere. These include the use of a class of molecules termed aptamers, which are chemically synthesized single strand nucleic acids, either RNA or DNA, that bind to target molecules with high selectivity and affinity leaving non-targeted protein functions intact.79,80 Other methods of direct inhibition of VEGF include inhibition of its tyrosine kinase receptors (VEGFR-1 and -2), either by systemic administration or gene transfer. Several laboratories have described the use of small-interfering RNA (siRNAs) in effectively downregulating gene regulation81 (see subsequent sections).
Other cytokines and regulators of angiogenesis
A number of cytokines have been identified that either upregulate VEGF or VEGF-associated effects, or act in an inhibitory capacity. In some instances, the same compounds may perform both functions depending upon the other extracellular signals present in the milieu as well as post-transcriptional or post-translational events, including cleavage by proteases, or binding by soluble receptors such as the family of tyrosine kinases.2 Accordingly, any of this class of normally occurring compounds can be considered either in their native form, through splice or cleavage products, or chemical substitution to be potential therapeutic compounds in the treatment of pathologic neovascularization. Examples of stimulatory cytokines in addition to vascular endothelial growth factor include basic and acidic fibroblast growth factor, the angiopoietins, transforming growth factor (TGF), IL-8, and platelet-derived growth factor (PDGF). Naturally occurring inhibitory factors include interferon alpha, thrombospondin 1, angiostatin, endostatin, hemopexin-like domain of matrix metalloproteinase 2 (PEX), and pigment epithelial-derived factor (PEDF).2,82–90 Additionally, it is likely there are many other naturally occurring compounds as well, as yet not recognized or well characterized biochemically. The relative balance between naturally occurring stimulatory and inhibitory factors is thought to contribute to the tight control of ocular vascular homeostasis, and may explain the differential ability of normal vascular as well as neovascular tissue to respond exquisitely both spatially and temporally to various stimuli.
Naturally occurring upregulators of angiogenesis
Fibroblast growth factor and integrins
Basic fibroblast growth factor (bFGF/FGF2) has been found to be elevated in excised choroidal neovascular membranes from patients undergoing surgery for AMD, and is capable of inducing pathologic neovascularization when implanted within the eye or adjacent to the retina.69,70 Laser models of choroidal neovascularization are accompanied by upregulation of bFGF-associated mRNA, which precedes the development of histologically identifiable new vessels. While bFGF is sufficient when inserted exogenously in creating choroidal neovascularization, it is not necessary; BFGF gene knockout animals are also capable of developing laser-induced CNV bFGF upregulation of angiogenesis is thought to be in part mediated by adhesion-mediated signals including expression of the cell membrane-associated integrins such as the alpha ν beta 3 integrin.2,69,70 Although the alpha ν beta 3 and another integrin, alpha ν beta 5, appear to differentially represent the stimulatory effects of bFGF and VEGF respectively in laboratory studies, there is conflicting evidence as to whether these pathways are distinct. As a result, considered therapeutic approaches to integrin inhibition may be directed at downstream events such as intracellular signaling in addition to targeted inhibition of the individual receptors through monoclonal antibodies or related mechanisms.
Platelet-derived growth factor
Platelet-derived growth factor (PDGF) is a homo- or heterodimeric glycoprotein that stimulates pericyte and vascular smooth muscle cell recruitment in angiogenesis, which are essential in the maintenance and survival of new vessels. The PDGF receptors (PDGFR-A and PDGFR-B) are tyrosine kinases, and are expressed differentially on different cell types, with PDGFR-B being more extensively expressed in endothelial cells, vascular smooth muscle cells, and fibroblasts, among others.91,92 In the absence of PDGFR-B signaling, vascular pericytes detach, and the integrity of vessel is lost. Immature vessels are highly sensitive to the withdrawal of VEGF prior to pericyte recruitment and coverage; conversely, when pericyte coverage is complete, new vessels are resistant to VEGF withdrawal. This is the so-called plasticity window, during which time vessels can regress rapidly with changes in exposure to growth factors.93,94 PDGF blockade in animal models of corneal neovascularization has resulted in prevention and regression of new vessels.95,96 PDGF blockade alone or combined with VEGF inhibition (taking advantage of the plasticity window) may thus serve as a potent inhibitor of neovascular AMD.
Angiopoietins
Angiopoietins, which are also highly specific for endothelial cells, perform a variety of other regulatory activities related to supporting cells and the extracellular matrix. A laboratory study suggests that two different isoforms, Angiopoietin 1 and Angiopoietin 2, appear to have differential and counteracting effects on the vasculature. Angiopoietin 2, which is upregulated by both hypoxia and VEGF, binds to an endothelial cell receptor TIE2 and enhances VEGF-mediated retinal neovascularization, but does not by itself stimulate endothelial cells or proliferation in vitro.2,97 Angiopoietin 1 appears to play a maturation role and is associated with nonleaky behavior and may have potential therapeutic benefit through its inhibition of inflammatory pathways.2
Matrix metalloproteinases and tissue inhibitors of metalloproteinases
Expression of matrix metalloproteinases (MMPs) and their associated RNA, particularly MMP-2 and MMP-9, have been associated with pathologic neovascularization. Both matrix metalloproteinases, which are bound to the integrin receptors alpha ν beta 3 and alpha ν beta 5 on endothelial cells, are activated by VEGF and other regulatory cytokines and act to digest the extracellular matrix and thereby facilitate the spreading and chemotaxis of actively proliferating endothelial cells as they aggregate to form new capillaries.2 Matrix metalloproteinases (MMPs) appear to be modulated and principally downregulated by naturally occurring tissue inhibitors of metalloproteinases (TIMPs) of which TIMP-1, TIMP-2, and TIMP-4 are thought to be soluble, and TIMP-3 principally extracellular matrix-bound. TIMP-3 is thought to play an important and possibly critical role in the natural modulation of matrix metalloproteinase regulation in Bruch’s membrane, and is found in increased amounts in drusen and thickened basement membranes associated with age-related macular degeneration.36,98 Mutations in exon-5 of the TIMP-3 molecule are associated with Sorsby’s fundus dystrophy,31,32,35 and an experimental model that either delivers increased amounts of TIMP-3 or induces overexpression of TIMP-3 by gene therapy demonstrates potent antiangiogenic effects for this molecule.99,100
Naturally occurring downregulators of angiogenesis
Pigment epithelial-derived factor
Pigment epithelial-derived factor (PEDF), which is secreted by the pigment epithelium in vivo as well as in cell culture, has been shown to be biochemically identical to the product of the wild type of retinoblastoma tumor suppressor gene (RB) and is thought to induce differentiation of retinoblastoma cells, inhibit microglial growth, and other important regulatory functions in addition to its effects on angiogenesis.56 Its relevance to AMD is likely related to its role in counterbalancing VEGF. In a study of donor human eyes with and without AMD, relative VEGF and PEDF levels were measured using immunoreactivity assays. Interestingly, VEGF levels were not significantly different between the two groups of eyes, even in the presence of advanced exudative change. In contrast, PEDF levels were much lower in the RPE and choroid of eyes with AMD, suggesting that it is lower levels of PEDF in the ocular milieu that may contribute to the formation of choroidal neovascularization.101 Results from in vitro studies have suggested that the role of PEDF is regulation or inhibition of VEGF receptor signaling, rather than direct VEGF antagonism.102
Other cytokines
Another interesting class of regulatory cytokines that appear to have an inhibitory effect on angiogenesis relate to the class of aminoacyl-tRNA synthetases, which are associated with the first step of protein synthesis. Tryptophanyl-tRNA synthetase (TrpRS, a homolog of TyrRS) has no effect in its native form on angiogenesis or angiogenesis signaling. In contrast, an alternatively spliced fragment reported to be stimulated by interferon-alpha and lacking a portion of the amino terminal fragment, has potent antiangiogenic effects in both in vitro and in vivo.103
Thrombospondin 1 (TSP-1) has been described as both an up- and downregulator of VEGF. Under specified conditions TSP-1, which is produced by platelets and monocytes, enhances simulated VEGF release and is dependent upon binding to alpha ν beta 5 and alpha ν beta 1 integrins. In separate studies, TSP-1 has also been noted to inhibit endothelial cell proliferation, migration, and angiogenesis. VEGF and TSP-1 appear to participate in a feedback loop and its potential role as a therapeutic agent remains uncertain.2 Angiostatin, is a 38 kDa internal fragment of plasminogen that encompasses the first four cringles of the molecule. It shows inhibitory effects on vascular endothelial proliferation in vitro and in vivo, particularly within tumors.90,104 Endostatin, a cleavage product of collagen XVIII, is structurally related to and shares homology with angiostatin, and inhibits tumor-associated angiogenesis.89 Each of these classes is further discussed in the following section as they relate to preclinical or clinical studies.
Agents currently in use or under investigation: non-neovascular AMD
Antioxidants, vitamins, and cofactors
The rationale for the use of antioxidants, multivitamins and other cofactors is based upon several lines of evidence, including the essential role of vitamin A in the visual transduction cycle, the known and predicted effects of oxidative stress resulting in reactive oxygen intermediates, and the requirement of certain critical metal ions including zinc in the functioning of critical proteases and other naturally occurring defense mechanisms against oxidative injury. Prior to the release of the data from the AREDS study, there remained some controversy as to whether antioxidant multivitamin therapy was efficacious, with some studies suggesting a protective effect, and others not.49,50,105
Age-Related Eye Disease Study (AREDS) and related supplements
The AREDS was a large, well-designed multicenter randomized clinical trial, which evaluated the effect of high-dose vitamin C and E, beta-carotene, and zinc supplements on AMD progression and visual acuity. A total 4757 persons were enrolled and stratified into one of four categories of increasing severity of disease. Due to the low rate of progression of category 1, consisting of drusen area of less than five small drusen of 63 µm or less, statistical analysis was only performed on the remaining 3640 patients with categories 2, 3, 4. Only the latter two (categories 3 and 4) were ultimately found to be at more than a trivial risk for progression. Patients in category 2 with at least one intermediate size druse, but not extensive drusen, had only a 1.3% probability of progression to advanced AMD by year 5 and no meaningful inference could be drawn regarding their risk with or without intervention. Patients in category 3 had either extensive intermediate drusen, large drusen, or non-central geographic atrophy, and patients in category 4 had visual acuity of less than 20/32 due to AMD in one eye related either to geographic atrophy involving the center of the macula or choroidal neovascularization in the fellow eye, but not the study eye, which had visual acuity 20/32 or better. The average follow-up of the 3640 enrolled study participants aged 55–80 was 6.3 years, and by comparison with placebo, patients treated with antioxidants plus zinc demonstrated a statistically significant odds reduction for the development of advanced AMD (OR 0.72, 99% CI 0.52–0.98). The odds ratio for zinc alone and antioxidants alone were 0.75 and 0.8, respectively, and the odds reduction estimates increased when only patients with category 3 or category 4 drusen were included, reducing to 0.66, 0.71, and 0.76 for antioxidants plus zinc, zinc alone, and antioxidants alone respectively. These data are further reflected in Fig. 67.9.
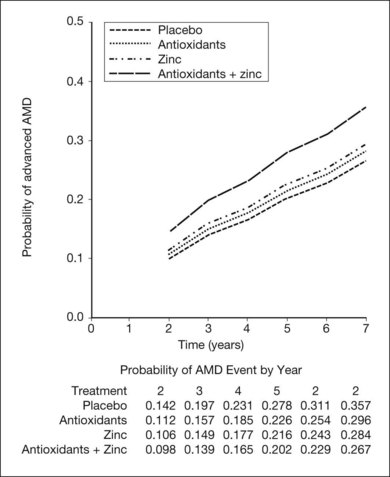
Based upon these findings, and extrapolation of the US population at risk, it has been estimated that of the 8 million persons in the United States at least age 55 or greater with AMD, if those at highest risk including categories 3 and 4 were to receive therapy with the formulation employed in the AREDS trial, as many as 300 000 of the 1.3 million at highest risk for advancement might avoid progression to the severe forms of the disease.106 Additionally, based upon recent epidemiologic data, exclusion of beta-carotene is recommended for persons with a current history of smoking, or a long smoking history based upon a theoretical increased risk of lung cancer.
The macular carotenoids thought to be the principal contributors to the yellow coloration of the macula include lutein and zeaxanthin. One study suggested that a high dietary intake of these carotenoids protected against AMD, resulting in a 43% reduction in risk for AMD when the upper quartile of use was compared with the lowest quartile.47 Recent AREDS reports have supported these findings, showing in cohort studies that those with a high level of dietary intake of lutein and zeaxanthin were less likely to have advanced AMD, as well as large or extensive intermediate drusen.107 High dietary intake of ω-3 long-chain polyunsaturated fatty acids were also found by the AREDS investigators to be inversely related to progression to both GA as well as neovascular AMD.44–46 The critical dependence of the naturally occurring antioxidant enzymes (superoxide dismutase, catalase, and glutathione peroxidase) as well as the matrix metalloproteinases, on copper, zinc and manganese, provides a potential mechanism by which AREDS supplementation of both copper and zinc could be beneficial. The naturally occurring antioxidants such as vitamin E and vitamin C might have a beneficial effect on the progression of AMD as well.48 At present no definitive recommendations can be made regarding dietary supplementation with lutein and zeaxanthin, as well as ω-3 long-chain polyunsaturated fatty acids. However, the AREDS2 randomized multicenter phase III clinical trial (NCT00345176) is currently underway, and is designed to address: (1) the role of lutein (10 mg)/zeaxanthin (2 mg) and ω-3 long-chain polyunsaturated fatty acids (350 mg docosahexaenoic acid (DHA)/650 mg eicosapentaenoic acid (EPA)) in the prevention of development of GA or CNV; and (2) the effect of possible deletion of β-carotene and lowering the daily zinc oxide dose from 80 mg to 25 mg. The AREDS study is discussed also in Chapter 65 (Age-related macular degeneration: Non-neovascular early AMD, intermediate AMD, and geographic atrophy).
Visual cycle inhibitors*
Visual cycle modulators are intended to reduce the accumulation of toxic fluorophores such as A2E in RPE cells. Fenretinide (ReVision Therapeutics, Inc.) treatment causes a dose-dependent, reversible reduction in circulating retinal binding protein (RBP) and retinol by displacing retinol from RBP, as well as interfering with RBP binding to transthyretin (TTR), thus facilitating their elimination via glomerular filtration. In ABCA4−/− mice, fenretinide reduces RPE lipofuscin and A2E accumulation, although it causes modest delays in dark adaptation.108 Notably, however, despite low blood retinol levels, RPB−/− mice acquire normal vision by 5 months of age when given a vitamin A-sufficient diet.109,110 Thus, it is not clear that blockade of vitamin A transport to RPE by inhibition of vitamin A binding to RBP will block vitamin A uptake by the RPE during long-term administration unless dietary vitamin A is restricted also. Patients in a phase IIb clinical trial of fenretinide received placebo, 100 mg, or a 300 mg daily dose for 24 months (NCT00429936). At the conclusion of the two-year study, an exploratory and ad hoc analysis of the data revealed that 15 (18.3%) of 82 patients in the placebo arm progressed to CNV, compared with 15 (9.2%) of 164 patients receiving fenretinide at either dose (P = 0.039). In the 300 mg fenretinide dose cohort, analysis of GA lesion growth by color fundus photography showed a trend for slowing of lesion growth, particularly among patients who had RBP and retinol levels reduced by more than 50%. Although fenretinide can affect dark adaptation111–118 and ERG readings,119–122 and is associated with symptoms of dry eye, 116,117 it was generally well tolerated in this study.
Accutane (13-cis-retinoic acid) inhibits the conversion of all-trans-retinyl esters (in retinosomes) to 11-cis-retinol and the conversion of 11-cis-retinol to 11-cis-retinal by retinol dehydrogenase and also reduces lipofuscin accumulation in ABCA4−/− mice.42,123 This oral agent is associated with a high incidence of nyctalopia.124 Another drug known as ACU-4429 (Acucela) is an orally administered compound that inhibits conversion of all-trans-retinyl ester to 11-cis-retinol via blockade of RPE65. ACU-4429 also reduces lipofuscin and A2E accumulation in the RPE of ABCA4−/− mice. A phase I clinical trial (NCT00942240) in 46 healthy volunteers was completed successfully.125 The most common adverse events were vision-related, occurring in 50% of patients receiving the medication, which included dyschromatopsia (32%), unspecified visual disturbance (29%), night blindness (18%), blurred vision (11%), and photophobia (8%). All adverse events were mild or moderate in intensity and were transient in nature, resolving within a few days after onset. There was dose-dependent suppression of the ERG b-wave as expected. A dose escalation phase II study is underway in patients with GA (NCT01002950).
Complement modulators*
In general, complement modulating agents work by replacing a defective complement component; by inhibiting the activation of convertases; by promoting the decay of convertases; or by blocking effector molecules (Fig. 67.10).126 The following examples illustrate some of the promise as well as the challenges associated with manipulation of the complement pathway.
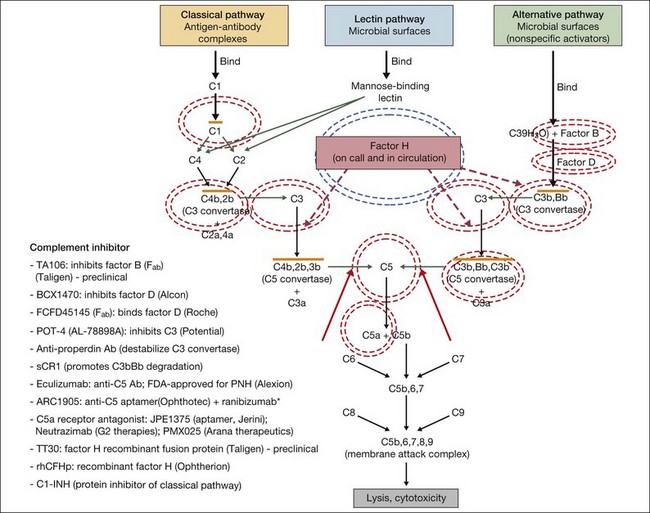
Fig. 67.10 Numerous compounds that modulate the complement pathway are in development for or in clinical trials for AMD treatment. Red circles indicate the parts of the complement pathway that are being modified. The complement pathway illustrated is adapted from Donoso et al.126
(Reproduced from Zarbin MA, Rosenfeld PJ. Pathway-based therapies for age-related macular degeneration: an integrated survey of emerging treatment alternatives. Retina 2010;30:1350–67 with permission of Springer Science+Business Media.)
Inhibition of C5 blocks terminal complement activity, but proximal complement functions remain intact, e.g., C3a anaphylatoxin production, C3b opsonization, and immune complex and apoptotic body clearance. ARC1905 (Ophthotech Corp.) is an anti-C5 aptamer delivered by intravitreal injection. It binds C5 with high affinity (KD = ~700 pM) and prevents cleavage to C5a and C5b. ARC1905 is in a phase I trial (NCT00950638) for patients with GA. Eculizumab (SOLIRIS, Alexion Pharmaceuticals) is a humanized monoclonal antibody that binds to and prevents cleavage of C5 and is administered intravenously. Eculizumab is FDA-approved for the treatment of paroxysmal nocturnal hemoglobinuria and is in phase II trials (NCT00935883) for treatment of nonexudative AMD, including patients with high-risk drusen or GA. C5a receptor blockade – e.g., JPE1375 (Jerini); PMX025 (Arana Therapeutics); Neutrazimab (G2 Therapies) – might have both advantages and disadvantages over direct C5a inhibition. C5a receptor blockade might inhibit some important inflammatory pathways,24 although without disabling membrane attack complex formation.
Factor D is the rate-limiting enzyme in the activation of the complement alternative pathway. It plays an important role in the positive feedback loop that results in the amplification of proinflammatory effectors.127 FCFD45145 (Genentech/Roche) is a monoclonal antibody fragment (Fab) directed against Factor D. A 108-patient placebo-controlled phase I clinical trial (NCT00973011) of intravitreal therapy for GA is complete, and the medication was well tolerated up to a 10 mg dose. Patients were treated monthly or every other month during an 18-month period. A phase II study (NCT01229215) is underway.
Replacement of complement factor H should inhibit inflammation in AMD patients with risk-enhancing mutations in CFH. This approach, which probably would require genetic screening prior to treatment, involves restoration of complement homeostasis so there is no increased risk of infection with therapy. Replacement of defective CFH is being developed by Alexion. TT30 is a recombinant fusion protein comprising complement receptor type 2 and the regulatory domain of factor H. The receptor domain of TT30 binds iC3b/C3d-coated cells, delivering the regulatory domain of CFH to areas where complement regulation is necessary.127 Preclinical testing is underway. Alexion is also exploring factor B inhibition using a humanized antibody fragment (TA106).
The differential expression of complement components in different ocular tissues presents difficulty in determining the optimal route of administration of these agents. For example, factor D is deposited diffusely in the human retina as well as in the choroid,128 while factors B, H, C3, C5, and C5b-9 are found primarily in the choroid, Bruch’s membrane, and/or subjacent to the RPE.128,129 Thus, intravitreal injection may not be the ideal route of delivery for all the complement inhibitors under study.
Another approach to complement modulation is the silencing of genes by preventing mRNA expression, as deletion of genes closely related to CFH (i.e., CFHR1 and CFHR3) appears to be strongly protective against AMD.130 However, short-interfering RNA (siRNA) therapies in the eye may be toxic, and it seems that the deletion of CFHR1 and CFHR3 protect against development of AMD at least in part because the deletion tags a protective haplotype and does not occur in association with the Y402H single nucleotide polymorphism.131 See below for further discussion of siRNAs and other complement inhibitors for exudative AMD.
Agents currently in use or under investigation: neovascular AMD
VEGF inhibitors
A variety of methods are now available to directly inhibit the VEGF165 molecule as well as its various other isoforms. These include the use of monoclonal antibodies directed at one or more isoforms, oligonucleotides with chain-specific sequences corresponding to the VEGF protein, and molecules directed at one or more of the VEGF receptors, including native decoys, tyrosine kinase inhibitors, fusion proteins, and TIE2 receptors. Finally, because VEGF is thought to initiate a cascade of intracellular signals initially, and subsequently extracellular events, it is possible to inhibit VEGF effects either through prevention of secretion of the molecule, direct inhibition of the molecule in the extracellular space, blockade of the receptors, or through interruption in the downstream intracellular signaling pathways leading to both intra- and extracellular events.132 In addition to the sections that follow, the interested reader is referred to Chapter 66 (Neovascular (exudative or “wet”) age-related macular degeneration).
Direct VEGF inhibitors
Monoclonal antibody: bevacizumab (Avastin)
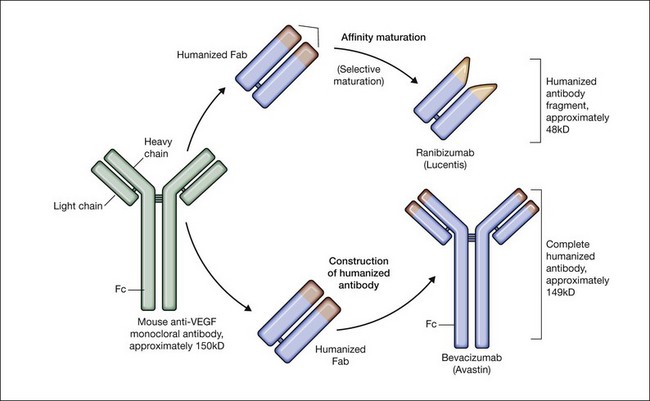
Bevacizumab is a humanized monoclonal antibody (IgG1) against human VEGF-A that selectively inhibits all isoforms and bioactive proteolytic breakdown products of VEGF-A (Fig. 67.11).133 Bevacizumab is an immunoglobulin G molecule that is comprised of amino acid sequences which are about 93% human and 7% murine. Preclinical studies in animal models of various tumor cell lines as well as different forms of ocular neovascularization indicated that the fully sized antibody had excellent efficacy against the primary permeability and proliferative effects of VEGF isoforms. Following extensive clinical testing, it was found to be effective in slowing tumor growth through its effects on angiogenesis.73 Bevacizumab was FDA-approved in 2004 as a treatment for metastatic colorectal cancer when given intravenously at a dose of 5 mg/kg and infused every 2 weeks in combination with 5-fluorouracil. Bevacizumab was shown to increase survival, response rate, and duration of response compared with conventional therapy consisting of irinotecan, fluorouracil, and leukovorin for metastatic colorectal cancer.134 Additional phase III clinical trials with bevacizumab have since resulted in FDA-approval for the treatment of lung, kidney, and brain cancers.
In 2004, a study investigating the use of systemic bevacizumab in patients with exudative AMD (Systemic Avastin for Neovascular AMD – SANA) was initiated at the Bascom Palmer Eye Institute. This study was the first demonstration that bevacizumab was effective for this disease. In this open-label, uncontrolled study, 18 patients underwent two to three intravenous infusions of bevacizumab (5 mg/kg) to evaluate the potential safety and efficacy of systemic bevacizumab over 6 months.135,136 Although clinical trials using intravitreal pegaptanib and ranibizumab for CNV were underway, the prior use of bevacizumab for ocular disease in humans using any route had previously been untested. While no serious adverse events were observed, and only a mild transient increase in blood pressure was identified, the SANA study was too small to establish the safety of bevacizumab. However, significant improvements in visual acuity and OCT central retinal thickness measurements were observed comparable to the changes observed in the early phase Lucentis trials.
While systemic bevacizumab (5 mg/kg) was shown to reduce leakage from CNV, decrease OCT central retinal thickness measurements, and significantly improve vision in exudative AMD, the intravenous use of bevacizumab for exudative AMD was never widely adopted, because the intravitreal approach used up to 500-fold less drug, is much less expensive, and is perceived to be safer due to the smaller dose of drug. In the first-reported case of intravitreal bevacizumab,137 a patient with recurrent CNV secondary to AMD, who had previously been treated with verteporfin photodynamic therapy (PDT) in combination with triamcinolone acetonide followed by treatment with pegaptanib injections, was shown to improve with a decrease in OCT central retinal thickness, subretinal fluid, as well as improvement of subjective visual distortion within 1 week of a single injection of 1.0 mg bevacizumab.
Initially, there was concern that a molecule as large as the 149 kDa bevacizumab may not penetrate the retina and effectively treat CNV compared with ranibizumab, which is one-third the size. However, it became apparent that bevacizumab could penetrate the retina based upon the empiric observations of decreased subretinal fluid on OCT examination and improved visual acuities in treated patients. Subsequently, Han et al. were the first to show that a full-length immunoglobulin the size of bevacizumab was capable of penetrating the rabbit retina after an intravitreal injection.138 Sharar et al. used qualitative immunofluorescence to confirm that intravitreal bevacizumab was able to completely penetrate the retina by 24 hours and was essentially absent at 4 weeks after an injection.139 Moreover, Dib et al. demonstrated bevacizumab molecules in the subretinal space of all six eyes studied two hours after an intravitreal bevacizumab injection of 0.05 ml (1.25 mg), confirming the initial observations that the molecule could rapidly diffuse through the retina.140
The ocular pharmacokinetics of various other monoclonal antibodies injected into the vitreous cavity of monkeys and rabbits demonstrated a half-life of about 5.6 days.141,142 Studies of bevacizumab in rabbits and monkeys suggest a half-life in the range of 4–6 days.143–145 A human study reported that a single dose of intravitreal bevacizumab had a half-life of 3 days and was likely to provide complete intravitreal VEGF blockade for a minimum of 4 weeks.146 One human study suggested a half-life of 6.7 days and yet another study has reported a half-life of as long as 9.8 days.147,148 It is likely that human ocular pharmacokinetics of bevacizumab varies from patient to patient, depending on factors such as the extent of vitreous liquefaction and the phakic status of the eye.
Since the first case of intravitreal bevacizumab for the treatment of exudative AMD was reported, numerous retrospective studies and a few small prospective studies using a dose range between 1.0 and 2.5 mg have been published, all demonstrating clinically significant improvement in mean visual acuity, reduction in fluorescein angiographic leakage, and resolution of OCT-visualized edema in up to 90% of bevacizumab-treated patients. These reports also supported the apparent clinical safety of bevacizumab. As of the time of writing (February 2012), despite the subsequent approval of ranibizumab in 2006, the off-label use of intravitreal bevacizumab has become the most common treatment for exudative AMD in the United State.149,150 In addition, it has also become frequently used for wide range of other forms of macular new vessels including pathologic high myopia, multifocal choroiditis, and ocular histoplasmosis; it has also been used successfully for treatment of clinically significant macular edema from diabetic retinopathy, retinal venous occlusions, and retinopathy of prematurity.151–159 It is being explored for treatment of other macular conditions not associated with new vessels or known elevated VEGF levels, including central serous choroidopathy and uveitides, although its efficacy in those conditions is not yet firmly established. Prior to the release of the Comparison of Age-Related Macular Degeneration Treatments Trial (CATT, discussed below), most of the early pilot studies were small (less than 100 patients) and uncontrolled, with different retreatment criteria and outcome measures; most physicians have frequently extrapolated the results from the phase III ranibizumab trials for these indications to bevacizumab.160 However, two randomized, controlled studies deserve mention.
The ABC trial compared intravitreal bevacizumab (three loading doses every 6 weeks, followed by additional injections at 6-week intervals as needed) to standard therapy, defined at the time of recruitment as verteporfin PDT for predominantly classic CNV, or pegaptanib injection or sham injection for minimally classic or occult CNV. A total of 131 patients were recruited and randomized 1 : 1 to receive either bevacizumab or standard therapy. A significantly higher proportion of the bevacizumab group improved by 15 letters compared with the standard therapy group (32% vs 3%, P < 0.001), irrespective of lesion type. Additionally, more patients lost fewer than 15 letters in the bevacizumab group (91% vs 67%, P < 0.001), and a larger proportion of bevacizumab patients achieved 6/12 or better vision by week 54 of the study. By study end, mean change in vision was +7.0 letters in the bevacizumab group, versus −9.4 letters in the standard care group. Serious ocular and nonocular adverse events were rare in all groups, with no serious intraocular infections in any of the intervention groups. Two myocardial infarctions, one fatal, occurred in the bevacizumab group.161 This study represented the first level 1 evidence of the efficacy of bevacizumab for the treatment of AMD.
A small randomized study conducted at the Veterans Affairs Boston Healthcare System Hospital compared bevacizumab and ranibizumab directly. A total of 28 patients were randomized in a 2 : 1 ratio to receive bevacizumab or ranibizumab, of which 22 patients completed one year of follow-up. Injections were given monthly for 3 months, followed by additional injections given on the basis of OCT findings primarily, or vision or clinical exam. Visual and anatomic results were similar between the two groups: patients receiving bevacizumab improved an average of 7.6 letters, compared with 6.3 letters in the ranibizumab group (P = 0.74). The change in central macular thickness as determined by OCT was −50 µm in the bevacizumab group, versus −91 µm in the ranibizumab group (P = 0.29). The number of ranibizumab injections administered was significantly lower than the number of bevacizumab injections (4 vs 8, P = 0.001). The authors hypothesized two possible reasons: (1) tachyphylaxis to bevacizumab; or (2) a more robust early anatomic response to ranibizumab, leading to a higher number of bevacizumab injections given on an as-needed basis determined primarily by OCT findings.162 This study, although small in size, was the first head-to-head trial of bevacizumab and ranibizumab, and demonstrated the similar efficacy of bevacizumab to ranibizumab.
Antigen binding fragment: ranibizumab (Lucentis)
Ranibizumab is a humanized anti-VEGF-A recombinant Fab fragment that has been affinity matured to increase its binding affinity for VEGF-A (see Fig. 67.11). Ranibizumab binds within the VEGFR-binding domain of all biologically active isoforms of VEGF-A. Two randomized, double-masked, pivotal phase III clinical trials have demonstrated that monthly intravitreal injections of ranibizumab are an effective and safe treatment for subfoveal CNV in AMD patients.163,164
The MARINA study assessed the response of minimally classic or occult CNV to ranibizumab.163 Patients (n = 716) were assigned randomly to receive sham injection (n = 238), 0.3 mg (n = 238), or 0.5 mg (n = 240) ranibizumab. After 4 months of follow-up, approximately 90% of ranibizumab-treated patients had lost less than 15 letters on the Bailey – Lovie (ETDRS) chart as compared to 53% of the sham-injected patients. This treatment response was independent of lesion size, initial visual acuity, or whether the lesion was classified as minimally classic or occult with no classic CNV on fluorescein angiography. Vision improved by at least 15 letters in approximately 33% of patients receiving 0.5 mg ranibizumab at 24 months of follow-up versus 4% in the sham-injected patients (Fig. 67.12).165 Approximately 1% of patients receiving intravitreal ranibizumab developed endophthalmitis. The risk of cataract was approximately 0.2%. There were no cases of retinal detachment among patients receiving intravitreal therapy. Despite the theoretical risk of systemic vascular complications, there was no imbalance among treated and control groups regarding hypertension, and the risk of myocardial infarction among sham-injected, 0.3 mg, and 0.5 mg cohorts was 1.7%, 2.5%, and 1.3%, respectively. The risk of stroke among the three groups was 0.8%, 1.3%, and 2.5%, respectively. The risk of nonocular hemorrhage was 5.5%, 9.2%, and 8.8% in each of the three cohorts, respectively. None of these differences was statistically significant, although the authors recognized that the study was not powered to detect small differences.
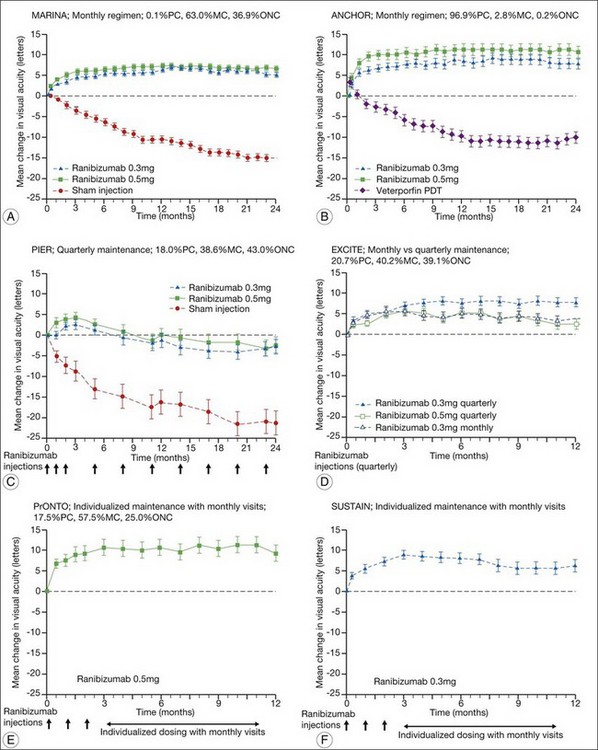
The ANCHOR study assessed the response of patients with predominantly classic CNV as determined by fluorescein angiography.164 Patients (n = 423) were randomly assigned to receive verteporfin-PDT plus sham injection (n = 143) or sham PDT plus injection of either 0.3 mg (n = 140) or 0.5 mg (n = 140) ranibizumab. At 12 months follow-up, approximately 95% of ranibizumab-treated patients lost less than 15 letters of vision versus 64% in the verteporfin active-treatment control group. Forty percent of patients treated with 0.5 mg ranibizumab gained at least 15 letters vision versus 6% in the verteporfin treatment cohort. At month 24, the visual benefit from ranibizumab remained statistically (P < 0.0001 vs PDT) and clinically significant: 89.9–90.0% of ranibizumab-treated patients had lost <15 letters from baseline (vs 65.7% of PDT patients); 34–41.0% had gained ≥15 letters (vs 6.3% of PDT group); and, on average, visual acuity was improved from baseline by 8.1–10.7 letters (versus a mean decline of 9.8 letters in the PDT groups).166 Changes in lesion anatomic characteristics on fluorescein angiography also favored ranibizumab (all comparisons P < 0.0001 vs PDT). Overall, there was no imbalance among groups in rates of serious ocular and nonocular adverse events. In the pooled ranibizumab groups, 3 (1.1%) of 277 patients developed presumed endophthalmitis in the study eye (rate per injection = 3/5921, or 0.05%). Retinal detachment was observed in one patient in both the verteporfin (0.7%) and 0.3 mg ranibizumab (0.7%) cohorts. There were no cases of lens injury. The risk of hypertension was the same in all cohorts. The risk of myocardial infarction among verteporfin, 0.3 mg ranibizumab, and 0.5 mg ranibizumab cohorts was 0.7%, 0.7%, and 2.1%, respectively. The risk of stroke or cerebral infarction was 0.7% in each of the three cohorts. The risk of nonocular hemorrhage was 2.1%, 5.1%, and 6.4% in each of the three cohorts. Immunoreactivity to ranibizumab before treatment was 1.5%, 3.2%, and 0.8% among the verteporfin, 0.3 mg ranibizumab, and 0.5 mg ranibizumab cohorts, respectively. Immunoreactivity was present in 1.6%, 1.6%, and 3.9% of patients in each of these cohorts, respectively, after 12 months of treatment. (The concern regarding immunoreactivity is that patients who develop an immune response might exhibit increased intraocular inflammation following intravitreal injection and consequently might not respond to the medication as well as patients who do not exhibit such a response. In practice, most retina surgeons have not observed this phenomenon and the drug has been minimally immunogenic in the doses tested.167)
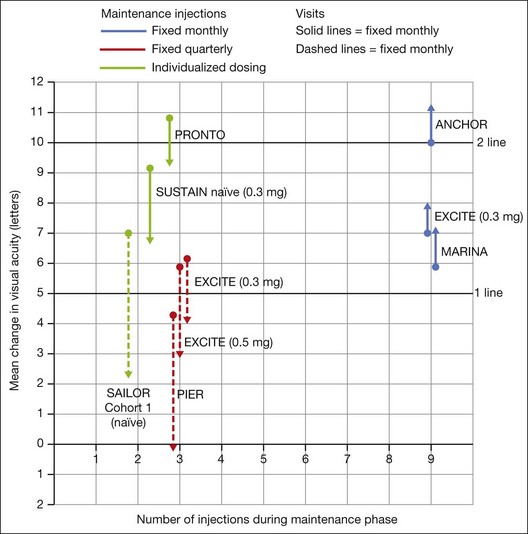
In both the MARINA and ANCHOR trials, visual acuity improvement seemed to reach a plateau by the 4-month time point (Fig. 67.12). Because monthly injections were considered inconvenient and daunting for some patients and their family members, and entailed risk as well as considerable expense, studies were undertaken to evaluate alternative treatment regimens. The PIER and EXCITE studies provided useful information in this regard.168,169 In PIER, a phase IIIb multicenter, randomized, double-masked trial, patients with subfoveal CNV were randomly assigned to sham injection (n = 63), 0.3 mg ranibizumab (n = 60), or 0.5 mg ranibizumab (n = 61). Patients received sham or ranibizumab injections every 4 weeks for three doses followed by additional treatment every 3 months. By month 12, mean changes from baseline visual acuity were −16.3, −1.6, and −0.2 letters for the sham, 0.3 mg, and 0.5 mg groups, respectively (P ≤ 0.0001, each ranibizumab dose vs sham). Ranibizumab arrested CNV growth and reduced leakage from CNV. The treatment effect declined, however, in the ranibizumab groups during quarterly dosing. At month 3, for example, the mean changes from baseline vision had been gains of +2.9 and +4.3 letters for the 0.3 mg and 0.5 mg doses, respectively. During study year 2, eligible sham-group patients crossed over to 0.5 mg ranibizumab quarterly. Later in year 2, all eligible randomized patients rolled over to 0.5 mg ranibizumab monthly dosing. The ranibizumab-treated patients showed an improvement in mean visual acuity during the first three months of the study, but this improvement was not sustained. Nonetheless, the 0.5 mg ranibizumab cohort had a mean visual acuity change that was 16 letters better than that of the sham cohort by month 12 (P < 0.0001). By month 24, visual acuity had decreased an average of 21.4, 2.2, and 2.3 letters from baseline in the sham, 0.3 mg, and 0.5 mg groups (P < 0.0001 for each ranibizumab group vs sham). The visual acuity of sham patients who crossed over to ranibizumab decreased over time, with an average loss of 3.5 letters 10 months after crossover. The visual acuity of patients in the 0.3 mg and 0.5 mg cohorts who rolled over to monthly ranibizumab injections increased for an average gain of 2.2 and 4.1 letters, respectively, 4 months after transition. These data indicate that injecting patients with ranibizumab every 3 months (after an induction phase of three monthly injections) does not produce the same chance for visual benefit as monthly injection, at least during the first 12 months of therapy. Ranibizumab appeared to provide additional visual benefit to treated patients who rolled over to monthly dosing, but not to patients who began receiving ranibizumab after >14 months of sham injections.
The EXCITE study, was a 12-month, multicenter, randomized, double-masked, active-controlled, phase IIIb study designed to demonstrate the noninferiority of a quarterly treatment versus monthly intravitreal ranibizumab treatment regimen in patients with AMD-associated CNV.170 Patients with primary or recurrent subfoveal CNV secondary to AMD (353 patients), including those with predominantly classic, minimally classic, or occult (no classic component) lesions, were enrolled. Patients were randomized (1 : 1 : 1) to 0.3 mg quarterly, 0.5 mg quarterly, or 0.3 mg monthly doses of ranibizumab. Treatment comprised three consecutive monthly injections followed by a 9-month maintenance phase (either monthly or quarterly injection). In the per-protocol population (293 patients), visual acuity increased from baseline to month 12 by 4.9, 3.8, and 8.3 letters in the 0.3 mg quarterly (104 patients), 0.5 mg quarterly (88 patients), and 0.3 mg monthly (101 patients) dosing groups, respectively. Similar results were observed in the intent-to-treat (ITT) population (353 patients). The mean decrease in CRT from baseline to month 12 in the ITT population was −96.0 µm in the 0.3 mg quarterly, −105.6 µm in the 0.5 mg quarterly, and −105.3 µm in the 0.3 mg monthly group. At month 12, the visual acuity gain in the monthly treatment cohort was higher than that of the quarterly regimens. The noninferiority of a quarterly regimen was not achieved with reference to 5.0 letters.
The SUSTAIN study is a 12-month, phase III, multicenter, open-label, single-arm study involving 513 ranibizumab-naive patients with AMD-associated CNV.171 In this study, patients received three initial monthly injections of ranibizumab (0.3 mg) and thereafter pro re nata (PRN) retreatment for 9 months based on prespecified retreatment criteria. Patients switched to 0.5 mg ranibizumab after approval in Europe. The average number of retreatments from months 3 to 11 was 2.7. Mean best-corrected visual acuity increased steadily from baseline to month 3 to reach +5.8 letters, decreased slightly from month 3 to 6, and remained stable from month 6 to 12, reaching +3.6 at month 12. The mean change in CRT was −101.1 µm from baseline to month 3 and −91.5 µm from baseline to month 12.
In summary, data from the ANCHOR, MARINA, PIER, EXCITE, and SUSTAIN studies indicated that after a loading dose period of three monthly injections, subsequently monthly ranibizumab injections give superior visual acuity outcomes compared to quarterly or PRN injections (Fig. 67.13).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


