Phakomatoses
Alex V. Levin
Thomas W. Wilson
Agnes Wong
The phakomatoses are a group of diseases that have cutaneous, central nervous system, and ocular manifestations. They often present with dermatologic and ocular manifestations, which subsequently lead to diagnosis of the underlying brain involvement. Significant vision loss can occur due to involvement of either the central nervous system or the eyes, or both.
At least three of the major phakomatoses—neurofibromatosis, tuberous sclerosis, and von Hippel-Lindau—share a common genetic basis: Mutation of a tumor suppressor gene leads to local cellular proliferations in the forms of tumor or skin changes. These three disorders are autosomal dominant in inheritance and demonstrate variable expression, though penetrance approaches 100%. As a result, the detection of affected family members may require extensive investigations, including neuroimaging and abdominal ultrasound. When counseling for autosomal dominant disorders, one must remember that even though each child of an affected parent carries a 50% risk of being affected, the severity of the disease may be very different between the parent and the child. Ataxia telangiectasia is the only autosomal recessive phakomatosis.
Some of the phakomatoses, such as Sturge-Weber syndrome, appear to be sporadic with only very rare reports of familial occurrence. This suggests a somatic (postzygotic) mutation resulting in localized affectation of the field of tissues that are the descendants of the mutated cell. Such disorders would only be heritable if the somatic change also affects the patient’s sperm or eggs. As the manifestations of these disorders are largely confined to the head and neck, gonadal involvement is uncommon.
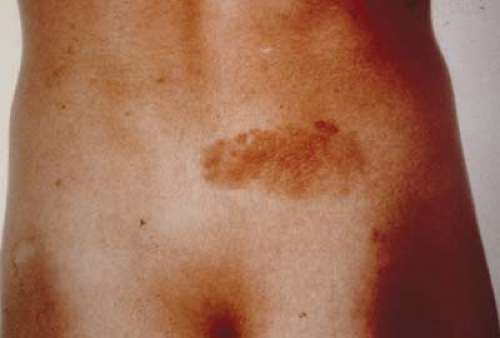 Figure 23.1 Tuberous Sclerosis—Shagreen Patch Tuberous sclerosis is an autosomal dominant phakomatosis caused by mutations in one of two genes: TSC1 at 9q34 or TSC2 at 16p13. The former produces the protein hamartin and the latter tuberin, both of which appear to have a role in cellular vesicular trafficking. Type 1 tuberous sclerosis has a lower incidence of mental retardation. There are no differences, however, in the ocular or dermatologic manifestations. Shagreen patches, shown here, are rough, raised lesions most commonly found in the lumbosacral region. They sometimes have an orange hue, but they may be more easily detectable by palpation than by inspection. |
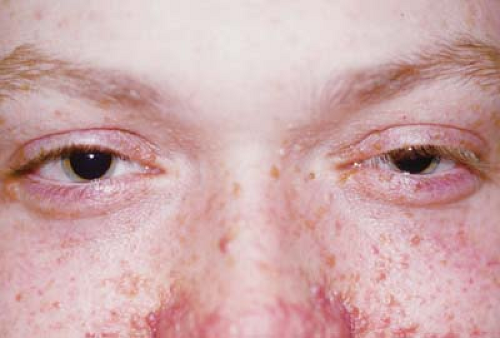 Figure 23.2 Tuberous Sclerosis—Adenomatous Sebaceum Adenoma sebacea are not true adenomas and are not of sebaceous origin. They are actually angiofibromas. The reddish lesions are distributed in a butterfly distribution on the face extending from the malar region across the bridge of the nose. They may be confused with the common cystic acne, particularly since the lesions can become more prominent during adolescence. This skin manifestation is rarely seen in the first decade of life. |
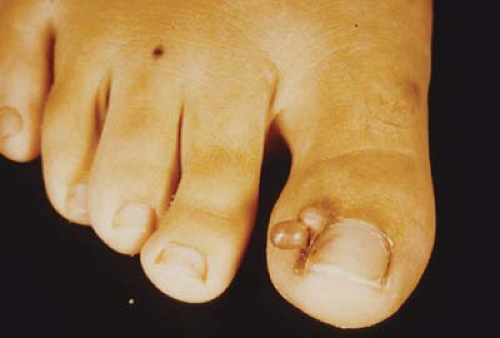 Figure 23.3 Tuberous Sclerosis—Subungual/ Periungual Fibroma Subungual/periungual fibromas are fibromatous growths under or around the nails, usually in the region of the cuticle. Despite their size, they are often asymptomatic but may require surgical intervention. They are rarely observed in the first decade of life and enlarge with age. |
 Figure 23.4 Tuberous Sclerosis—Ash Leaf Spot Ash leaf spots are hypopigmented lesions of the skin that often have edges and a shape reminiscent of an ash tree leaf. They are sometimes difficult to detect, especially in children with fair skin. However, they can be easily visualized with a Wood ultraviolet light. One must be careful to ensure that a false-positive Wood lamp test is not being caused by debris and other substances, which will also illuminate. The melanosomes in the region of the lesion are abnormally small and contain less melanin than the surrounding skin. The number of melanocytes is normal. |
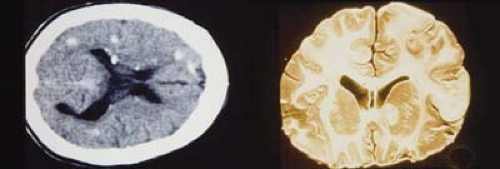 Figure 23.5 Tuberous Sclerosis—Cortical Lesions Central nervous system manifestations of tuberous sclerosis include seizures, mental retardation, astrocytic hamartomas of the brain (cortical tubers, shown here), and subependymal calcifications. Seizures are typically myoclonic spasms that present in infancy and occur in many patients with tuberous sclerosis. As shown in these images, the benign astrocytic hamartomas often appear as calcified lesions and are most commonly located in the periventricular area. Neurosurgical intervention is rarely needed and visual field defects from the tubers are also uncommon. |
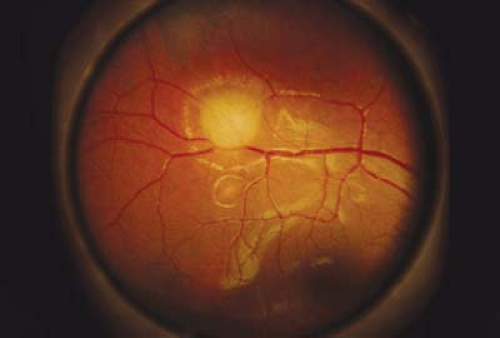 Figure 23.6 Tuberous Sclerosis— Retinal Astrocytic Hamartoma Retinal astrocytic hamartomas are one of the cardinal signs of the disorder. The sessile lesions are located in the nerve fiber layer and, as shown here in an infant, typically begin as noncalcified lesions. Note that the lesion is almost translucent, and in some cases, it may be difficult to recognize as it may only start as a faint gray superficial patch. The lesions have a predilection for the posterior pole but can occur anywhere in the retina. They are benign and usually of no visual consequence. |
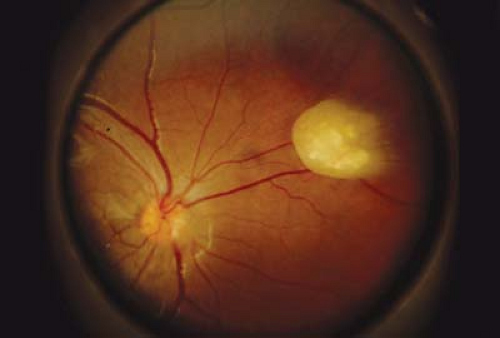 Figure 23.7 Tuberous Sclerosis— Retinal Astrocytic Hamartoma As the lesion matures, it becomes more opaque and eventually calcifies. In all phases, the differential diagnosis must include retinoblastoma (Chapter 8: Retina and Vitreous, Figs. 8.42, 8.43, 8.44, 8.45, 8.46, 8.47, 8.48, 8.49, 8.50, 8.51, 8.52 and 8.53). It can be differentiated from retinoblastoma based on its smooth surface, the absence of associated vascular abnormalities, exophytic growth, or vitreous seeding; and the presence of other systemic manifestations. Eyes have unfortunately been enucleated due to an incorrect suspicion of retinoblastoma. |
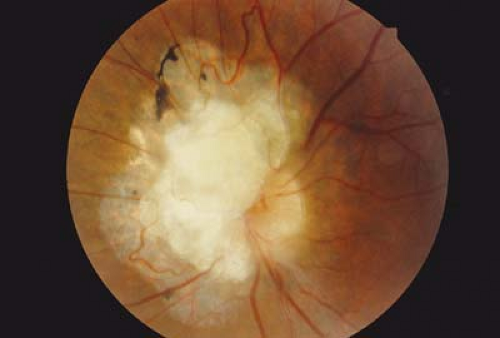 Figure 23.8 Tuberous Sclerosis—Optic Nerve Hamartoma Astrocytic hamartomas of tuberous sclerosis also have a predilection to involve the optic nerve head. The risk of leakage resulting in an exudative retinal detachment or hemorrhage, but still quite low. This photograph shows the remaining calcified mass following retinal laser to eliminate the exudative tumor. The lesions initially appear to have a multilobulated surface with intralesional calcium that may also be confused with retinoblastoma. The nickname “mulberry lesion” is sometimes applied. |
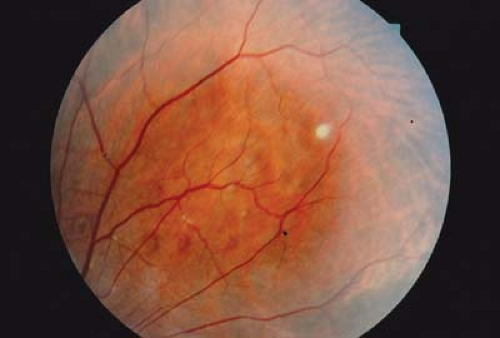 Figure 23.9 Tuberous Sclerosis—Retinal Pigmentary Lesion Areas of depigmentation may occur in the retinal periphery or midperiphery. The discrete lesion represents an area of decreased melanosome size and decreased melanin concentration similar to ash leaf spots (Fig. 23.4) of the skin. The lesion involves the retinal pigmented epithelium. It is benign, may be multiple, usually does not have an ash leaf mulberry appearance, and does not involve the overlying sensory retina. These lesions are visually insignificant. They are not diagnostic of tuberous sclerosis, as small hypopigmented lesions can be due to a variety of causes and may even be seen in normal children. |
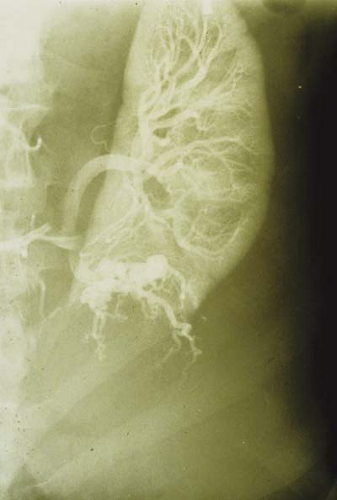 Figure 23.10 Tuberous Sclerosis—Renal Cyst
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|