Skeletal
Alex V. Levin
Thomas W. Wilson
Skeletal and connective tissue disorders will often have significant ocular manifestations. The connective tissue disorders are often the result of defects within tissue collagen. The abnormal collagen leads to structural weaknesses of the cornea, sclera, lens zonules, and Bruch membrane. These defects also cause systemic disorders of the skin, musculoskeletal system, heart, and vascular system. Collagen abnormalities of the cornea result in keratoconus and keratoglobus (Chapter 5: Cornea, Figures 5.18 through 5.20). Structural defects within the sclera lead to globe elongation resulting in high myopia, retinal detachment, and susceptibility to ruptured globe following minor trauma. Weakness of the Bruch membrane (the basement membrane of the retinal pigment epithelium) results in angioid streaks and subsequent vision-threatening subretinal neovascular membrane. Ectopia lentis is secondary to abnormal lens zonules. Vitreous collagen abnormality may lead to retinal detachment.
Similar defects in collagen may affect the heart valves and vascular tissues. Patients may present with an ocular problem and have a life-threatening systemic abnormality. One example would include a patient with Marfan syndrome presenting with symptoms of ectopia lentis and aortic root dilation detected before aneurysmal rupturing. A patient presenting with acute changes in vision secondary to angioid streaks and subretinal neovascular membrane may have the potential for severe uterine or gastrointestinal bleeding. Ocular abnormalities may be the key factor in the identification of a skeletal system disorder that also has other associated serious systemic abnormalities.
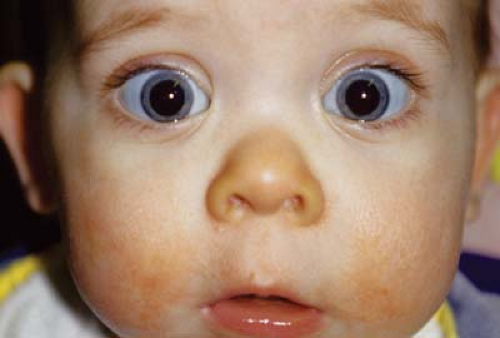 Figure 28.1 Stickler Syndrome Stickler syndrome is an autosomal dominant–inherited disorder with ocular and systemic features. The underlying abnormality is in the production of type 2A1 (12q13.11), 11A1 (1p21), or 11A2 (6p21.3) collagen. The latter form does not have ocular signs. Clinical features include high myopia, wedge-shaped cortical cataract, optically empty vitreous, perivascular lattice, and retinal detachment. Systemic findings include midfacial hypoplasia with Pierre Robin sequence, deafness, heart valve abnormalities, and progressive arthropathy. This photograph demonstrates pseudoproptosis from the axial myopia as well as the characteristic facies with midface hypoplasia and flat nasal bridge. |
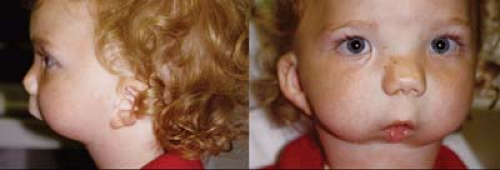 Figure 28.2 Pierre Robin Sequence The Pierre Robin sequence is also associated with Stickler syndrome. This complex includes micrognathia, high arched or cleft palate, and relative glossoptosis. The primary defect is the failure of the fetal mandible to grow. This leaves the tongue in an elevated position, which prevents closure of the palatal shelves. Approximately one-third of patients with Pierre Robin sequence have no systemic manifestations, one third have a recognizable syndrome, and one third have other manifestations but no recognizable syndrome. The ophthalmologist must be careful when examining children with Pierre Robin sequence in the supine position, as the tongue may obstruct the airway. In this child, recurrent tongue obstruction led to the need for tracheostomy. |
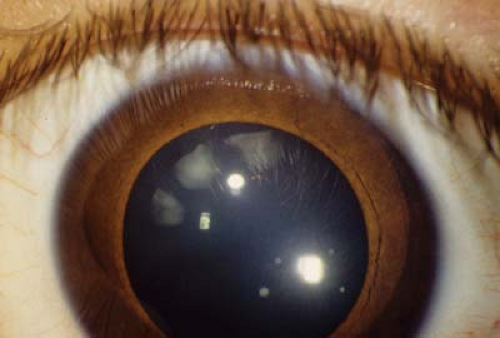 Figure 28.3 Stickler Syndrome— Cortical Wedge Cataract Cataracts are common in patients with Stickler syndrome, but due to the peripheral location, they may not require surgery. However, more visually significant cataracts may occur including presenile nuclear sclerosis and even total white cataract. The cataract in this photograph is the classic wedge-shaped cataract and is not causing any visual loss. Due to the vitreous abnormalities in this syndrome and the high risk for retinal detachment, it is advisable not to invade the posterior capsule during surgery. Fortunately, cataract surgery is rarely needed in infancy. |
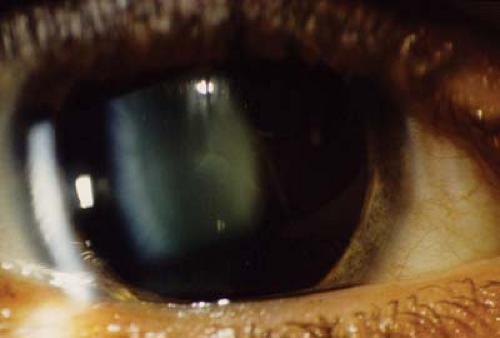 Figure 28.4 Stickler Syndrome— Vitreous Abnormalities The vitreous of patients with Stickler syndrome becomes optically empty. The vitreous is liquefied but may contain freely mobile avascular bands or veils. These bands do not cause significant retinal traction or visual compromise. In Stickler syndrome due to mutations in the COL11A1 gene, the vitreous may be visible but synergetic with beaded fibrils. In this photograph, the vitreous can be seen in the retrolenticular space and fleck-shaped opacities are located with the lens. |
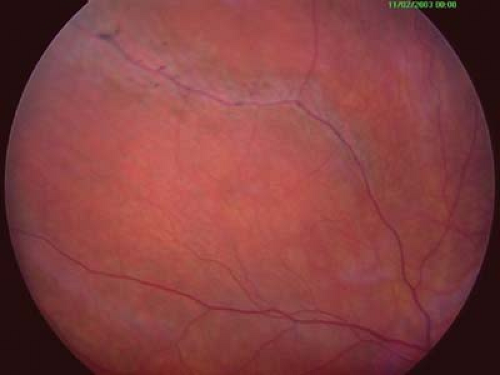 Figure 28.5 Stickler Syndrome— Perivascular Lattice The retina of Stickler syndrome is atrophic and is typical of high myopia. The refractive error is typically 8 to 12 diopters and nonprogressive. Ametropic amblyopia can occur if not detected at an early age. Perivascular lattice degeneration, shown here, in the peripheral retina and breaks within the retina are common. Children should be monitored on a regular basis with a peripheral retinal examination because of the high risk of retinal detachment. Stickler syndrome is the most common systemic disorder associated with giant retinal tears in children.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|