NEUROOPHTHALMIC DISORDERS IN the pediatric population, although not as common as in their adult counterparts, are equally crucial to patients’ ultimate visual and neurologic health. Very subtle initial symptoms may indicate major intracranial pathology. Prompt diagnosis and management is the best method to prevent permanent visual and neurologic sequelae.
The diseases encountered in pediatric neuroophthalmology are similar to those in adult situations in that the same broad categories like neoplastic, infectious, inflammatory, vascular, metabolic, and congenital must be considered. However, in pediatric neuroophthalmology, the frequency of the various diseases is weighted toward congenital, postinfectious, and neoplastic entities.
The clinician evaluating these patients must define the onset and progression of the present problem, and secure detailed prenatal, birth, growth and development, and family histories. The amount of history obtained directly from the child obviously depends on his or her age and mental status. Primary and secondary historical details must always be elicited from mothers, fathers, grandparents, and siblings who may have witnessed the patient’s behavior.
In this chapter, pediatric neuroophthalmic problems are approached as the patients and their parents report them to clinicians by their symptoms. Each symptom complex is analyzed anatomically so that a coherent comprehensive differential diagnostic approach may be formulated.
VISUAL LOSS
Children may not complain of loss of vision (especially if mild or unilateral), and usually adapt to the deficit rather than admitting to any interference with their activities. A decrease of visual acuity in one or both eyes is often detected on routine screening examinations performed by pediatricians, school nurses, and general ophthalmologists. After the more common problems of refractive error and strabismic or anisometropic amblyopia have been excluded, and if no anterior segment pathology can be found, concern must arise that a neuroophthalmic disorder is present. The optic nerve is the initial neuroanatomic structure of the afferent visual system and should occupy the first position in the differential diagnostic analysis.
The physical findings of unilateral optic nerve disease are identical for children and adults including decreased central visual acuity often associated with an optic nerve type visual field defect, an acquired dyschromatopsia, and a relative afferent pupillary defect (RAPD) or Marcus Gunn pupil depending on the severity of the optic neuropathy.
Bilateral optic neuropathies in some ways present a more difficult diagnostic challenge as the RAPD may be minimal or absent. Also, with bilateral optic neuropathies, dyschromatopsias may be symmetric, mimicking congenital color blindness. Finally, with bilateral, symmetric disease, the only pupillary finding is a sluggish reaction to direct light stimulation and a better near reaction, which is more difficult to appreciate than a RAPD.
Once the diagnosis of a unilateral or bilateral optic neuropathy has been established, the exact etiology must be determined. Optic nerve abnormalities can be isolated, associated with other ocular pathologies disorders of the central nervous system (CNS), part of a systemic disease, or syndromes. Toxic exposure during pregnancy (e.g., medications, illicit drugs, alcohol) and systemic maternal disorders (e.g., diabetes, seizure disorder) should be sought in the history. In addition, congenital defects of the optic nerve should be considered high in the differential diagnosis.
Optic nerve hypoplasia may present to the pediatric ophthalmologist during the evaluation of unilateral or bilateral visual loss or during the assessment of strabismus or nystagmus. Sometimes the patient is referred for further evaluation because of the unusual appearance of the optic disk. This disk anomaly may be the harbinger of associated significant CNS and endocrinologic abnormalities.
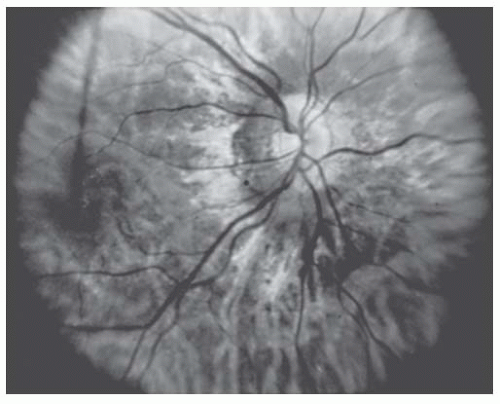
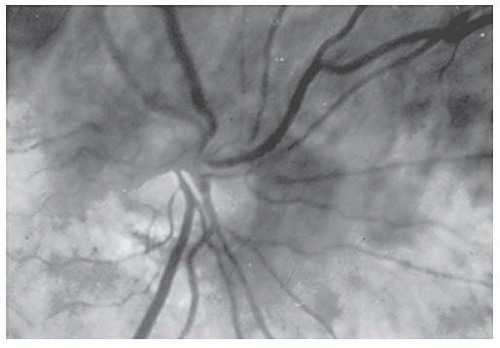
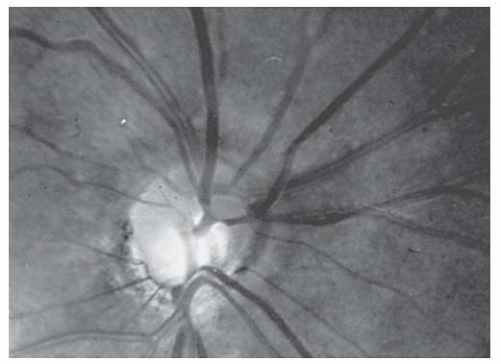
Hypoplastic optic nerves may be unilateral or bilateral, may affect central visual acuity minimally or profoundly, and may produce any nerve fiber type visual field changes ranging from nasal defects to more typical nerve fiber layer defects, diffuse depression, or generalized constriction (1). When the optic nerve is strikingly hypoplastic (Fig. 22.1), the fundoscopic diagnosis is relatively easy. However, detailed evaluation of the optic nerve with higher magnification may be necessary to evaluate subtle findings of optic nerve hypoplasia (Fig. 22.2).
FIGURE 22.1. Marked optic disk hypoplasia. The optic disk is approximately one-half the normal size and is surrounded by a peripapillary yellowish border.
Six fundoscopic features (Table 22.1) should be sought in optic nerve hypoplasia. First, by definition, the disk must be small. Second, the disk is typically surrounded by a peripapillary yellowish border that encircles the entire disk to form a halo. This halo constitutes the “double-ring” sign that has been invariably associated with this diagnosis. On the basis of the histopathologic study of Mosier and colleagues (2), the outer portion of the double-ring is the junction between sclera and lamina cribrosa where the choroid is discontinuous. The inner ring is demarcated by termination of the retinal pigment epithelium, with the whitish appearance of the inner ring attributed to glial and connective tissue around the retinal vessels.
FIGURE 22.2. More subtle hypoplasia of the optic disk than shown in Figure 22.1. The lower border of the disk is truncated. The patient presented with a superior visual field defect that was initially believed to represent a chiasmal lesion.
Table 22.1 FUNDOSCOPIC CRITERIA FOR DIAGNOSIS OF OPTIC NERVE HYPOPLASIA
Small optic disk
Peripapillary halo surrounds disk (“double-ring” sign)
Combined size of small disk and halo roughly approximates size of a normal disk
Normal or slightly tortuous, nondilated vessels
Decreased foveal reflex
Decreased thickness of retinal nerve fiber layer
Adapted from Rangwala LM, Liu GT. Pediatric idiopathic intracranial hypertension. Surv Ophthalmol 2007;52(6):597-617.
The third criterion for the fundoscopic diagnosis is that the dimensions of the hypoplastic nerve and peripapillary halo roughly approximate the size of a normal optic disk. The remaining three criteria involve the retina and include (a) normal or slightly tortuous, nondilated retinal vessels; (b) decreased foveal reflex; and (c) decreased thickness of the retinal nerve fiber layer. Pathologic studies have revealed decreased numbers of retinal ganglion cells, as well as thinning of the nerve fiber layer, thereby confirming the ophthalmoscopic observations of the retina (3).
It is important to remember that the appearance of the optic nerve is not always predictive of the ultimate visual acuity and can range from 20/20 to no light perception (4). In unilateral or asymmetric disease, there can be a superimposed component of amblyopia which may be amenable to conventional amblyopia treatment (5,6).
Etiology
No single, unifying pathophysiologic concept has evolved to explain optic nerve hypoplasia, up to 45% of optic nerve hypoplasia are sporadic but many environmental factors have been reported associated with optic nerve hypoplasia including maternal insulin-dependent diabetes mellitus, epilepsy, and younger maternal age, and the use of a variety of therapeutic and “recreational” drugs including lysergic acid diethylamide and ethanol (fetal alcohol syndrome) (7). In the therapeutic category, anticonvulsants, corticosteroids, diuretics, isoretinoin, meperidine (4,8,9,10), protamine, zinc, and insulin have all been reported to be associated with this optic nerve abnormality (11). Histologically, the number of optic nerve axons is reduced with normal supporting glial tissue. One proposed pathogenesis is supranormal regression of optic nerve axons in utero rather than a primary failure of differentiation (4,12,13). Recent genetic studies have implicated the homeobox gene HESX1/hesx1 in septooptic dysplasia. The data suggest that this gene plays an important role in forebrain and pituitary development (14,15,16,17).
Ocular and Central Nervous System Associations
Optic nerve hypoplasia may exist as a totally isolated congenital defect, but it has also been reported with almost every other congenital ocular abnormality ranging from ocular motor palsies to microphthalmos to blepharophimosis. Moreover, a variety of neurologic abnormalities and syndromes have been associated with hypoplastic optic nerves. Some of these disorders, such as hydranencephaly and anencephaly, preclude continued growth and development.
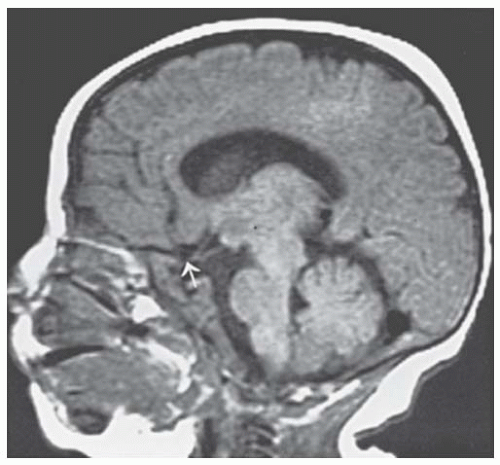
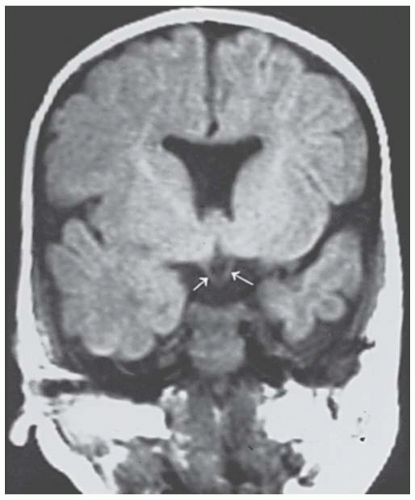
The septo-optic dysplasia or de Morsier (18,19) syndrome has been described as optic nerve hypoplasia, brain midline defects including agenesis of the anterior commissure and septum pellucidum, as well as malformation of the chiasm (Figs. 22.3 and 22.4), and pituitary hormone abnormalities (20). de Morsier coined the term “septo-optic dysplasia” for these defects, but it was Hoyt and coworkers (21) who stressed the syndrome of septo-optic dysplasia with growth retardation secondary to hypopituitarism. Magnetic resonance imaging (MRI) of the brain and orbits should be considered in patients with optic nerve hypoplasia (22). Detection of this syndrome is critical because the endocrine abnormalities can be life threatening. In addition, growth retardation is reversible if growth hormone therapy is initiated before epiphyses close. Likewise, encephaloceles are more common in these patients, and this brain tissue in the sphenoid sinus might be mistaken for an abnormality and even inadvertently biopsied.
FIGURE 22.3. Sagittal magnetic resonance imaging (MRI) scan of a patient with de Morsier syndrome (septooptic dysplasia). The pertinent neuroradiologic features are hypoplastic optic nerves (arrow), as well as agenesis of the anterior commissure and septum pellucidum (courtesy of Dr. Robert Grossman).
FIGURE 22.4. Coronal magnetic resonance imaging (MRI) scan of the patient in Figure 22.3. Arrows denote hypoplastic optic nerves.
OPTIC NERVE DYSPLASIA
Dysplasia of the optic nerve refers to a collection of clinical entities ranging from the morning glory syndrome and optic disk coloboma and other cavitary disk anomalies to megalopapillae and tilted optic disks. Of these, the morning glory syndrome and disk coloboma are discussed here because they may produce central visual loss in the pediatric-age group. The other disorders usually are associated with visual field defects and are discussed in the section concerning visual field abnormalities, but it is well known that tilted optic disks can produce a pseudobitemporal hemianopsia.
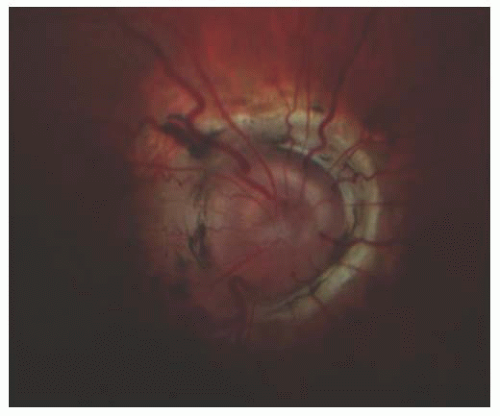
The morning glory syndrome (Fig. 22.5) is a rare disk anomaly first described by Kindler (23). It is comprised of a funnel-shaped excavation of the optic disk, distorted disk architecture, and a larger size than normal. The disk is surrounded by elevated, prominent, chorioretinal pigment in the shape of an annulus. There is typically a white tuft of glial tissue over the central portion of the disk. The blood vessels are a nomalous and arise from the periphery of the disk and often branch at acute angles. The term reflects the morphological similarity to the flower of the morning glory plant. The pathogenesis of the condition is unknown but there are different hypotheses including failure of closure of the fetal fissure or primary mesenchymal abnormality or dilation due to dysgenesis of the terminal optic stalk. Patients can present with transient visual loss episodes or chronic visual loss. An anomalous communication between the subretinal and subarachnoid space permits fluid to produce serous retinal detachments. Patients (24,25) often are neurologically and ophthalmologically normal but basal encephaloceles have been associated with the morning glory syndrome.
FIGURE 22.5. Morning glory syndrome of the optic disk. The disk is funnel shaped, distorted, and larger than normal size. The chorioretinal pigment surrounding the disk is shaped like an annulus.
Patients with this syndrome may lose vision because of both serous and rhegmatogenous retinal detachments (25). The prognosis for visual rehabilitation with traditional vitreoretinal surgical procedures is poor. However, Irvine and coworkers (26) and Chang and associates (27) reported some success in repairing these detachments when optic nerve sheath decompression surgery was performed simultaneously with vitreoretinal surgery.
Coloboma is the other major variety of dysplastic optic disk anomaly that may present in childhood with visual loss (Fig. 22.6). A coloboma is a developmental malformation with bowl-shaped excavation of the optic disk, which is deeper inferiorly resulting from partial or anomalous closure of the two sides of the proximal end of the embryonic fissure. An optic nerve coloboma may exist independently or be associated with colobomatous defects in the retina and choroid, depending on which portion of the embryonic fissure fails to close. If the proximal portion of the fissure fails to close in the correct manner, the retina and choroid are involved primarily. When the more distal segment of the fissure remains open, the nerve and its meningeal sheath are affected with a colobomatous defect. Optic nerve coloboma may occur sporadically or be inherited in an autosomal dominant pattern. It has been shown to be associated with PAX2 gene mutations as part of the renal-coloboma syndrome (25,28,29,30,31).
FIGURE 22.6. Optic disk coloboma, which was not associated with a corresponding colobomatous defect in the retina and choroid.
Patients with optic disk coloboma frequently have reduced visual acuity but the only feature that relates to visual outcome is the degree of foveal involvement by the coloboma. The visual field defects in coloboma however are variable. Significant refractive errors and anisometropia are also common in optic nerve coloboma and should be addressed (32,33). Patients with optic nerve coloboma are also at risk for retinal detachment, which has been reported to occur as early as 5 months of age (34). Peripapillary choroidal neovascularization has been described too (35).
As in optic nerve hypoplasia, optic nerve coloboma may occur in isolation or associated with other neurologic or systemic syndromes. Coloboma are most notably seen in CHARGE (coloboma, heart disease, atresia choanae, retarded growth and development, genitourinary abnormalities, and ear abnormalities) association and renal-coloboma syndrome (36).
Early intervention is essential for children with poor visual acuity due to any of these conditions, and these conditions are managed by treating refractive errors, occlusion therapy, and optimizing the conditions at home and at school in an attempt to ensure that impaired vision does not impede development or education (25).
HEREDITARY OPTIC ATROPHY: DOMINANT OPTIC ATROPHY
Dominant optic atrophy (DOA) is the most common bilateral hereditary condition of the optic nerve encountered in the pediatric population. The prevalence of DOA is 1 in 35,000 individuals in northern Europe. Recent studies have shown that mutations in the nuclear-encoded dynamin-like GTPase nuclear gene OPA1, which is involved in mitochondrial fusion, are responsible for the majority of DOA cases. The OPA1 gene causes DOA linked to chromosome 3q27-q29 (37,38,39,40). Patients often present in childhood after failing school screening examinations. Acute visual loss has not been described in this disease. Some patients may not come to medical attention until adulthood. A patient with a DOA mutation has a 50% probability of transmitting the pathogenic allele to each of the siblings. DOA demonstrates incomplete penetrance and children with the mutant allele have a 66% to 88% chance of manifesting the disease (41,42).
Because the visual deficits may be mild (20/40 to 20/200 range), visual acuity typically decreases over the first two decades of life. Thinning of the neuroretinal rim is a universal finding (43,44) and because children frequently do not report visual changes, the exact incidence and onset of DOA remains uncertain. Classically, DOA is detected during primary school vision screening (45) and 58% to 84% of patients report visual impairment by age of 11 years (46,47).
The degree of vision loss varies among the same family members and could be asymmetric in affected individuals. Although most patients with DOA had isolated optic neuropathy, up to 20% are associated with neurological manifestations such as sensorineural hearing, myopathy, peripheral neuropathy, ataxia, ptosis, and/or ophthalmoplegia (48,49).
In 1959, Kjer (50) published his classic monograph describing 19 Danish families with DOA. He attempted to use the presence of nystagmus as a distinguishing feature between what he believed were two forms of DOA. However, Waardenburg and coworkers (51) considered that the nystagmus was too nonspecific a finding to be used reliably to separate these two supposed entities. Review of the literature favors the position of Waardenburg. Kline and Glaser (52) described 24 individuals in 4 pedigrees without nystagmus or other extraocular motility abnormalities.
This study may represent the best description of the clinical profile of DOA. In this series, best corrected visual acuity ranged from 20/25 to 20/400. In 6 of the 12 patients, the two eyes had identical acuity, and there was a one line difference between the two eyes in two patients. The remaining four patients had greater than a two-line difference between eyes, one individual having 20/30 in the right and 20/200 in the left.
Visual field deficits for DOA are noteworthy for four characteristics: (a) elongated blind spots, (b) cecocentral scotoma, (c) mild temporal depression to central isopters (I-2-E and I-3-E) are rarely encountered, and (d) normal peripheral isopters (I-4-E or larger). Progressive loss of acuity and visual field have been described rarely in DOA. Such deterioration is so unusual that thorough diagnostic investigation is advocated in this circumstance, so that “progressive DOA” becomes a diagnosis of exclusion.
The color vision deficits in DOA are fascinating and often provide a helpful diagnostic point in distinguishing DOA from other optic neuropathies. Ishihara and Hardy-Rand-Rittler provide excellent screening methods for detecting dyschromatopsias for DOA, but only the Farnsworth-Munsell 100 hue examination precisely defines the defects. In Kline and Glaser’s (52) series, a tritan (yellow-blue) error was found in 15 of 22 eyes. In 5 of these 15 eyes, a deutan or protan axis was also identified with the tritan abnormality. The remaining seven eyes displayed a generalized dyschromatopsia without an identifiable axis. This tritan dyschromatopsia for DOA is significant because it violates Koellner’s rule (i.e., disease of receptor and bipolar layer of the retina results in decreased blue-yellow sensitivity and disease of the retinal ganglion cells and visual pathways anterior to the lateral geniculate body produces red-green dyschromatopsia). To date, no explanation for the unusual tritan axis in DOA has been provided.
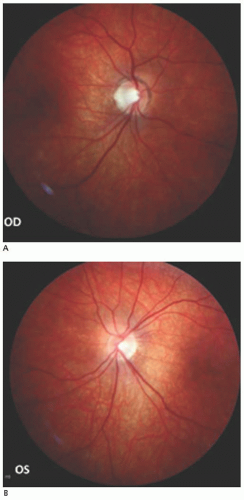
The appearance of the optic nerve in DOA has been controversial. There is agreement that pallor of the temporal segment of the disk occurs routinely and that diffuse pallor does not occur, but no consensus has been reached regarding other features of the optic nerve appearance. Kline and Glaser (52) believed strongly that most eyes (16 of 22 in their series) demonstrate focal temporal excavation (Fig. 22.7). This excavation can be so pronounced that the temporal portion of the disk may be on a different plane compared with the nasal aspect. The degree of temporal excavation is highly variable, and this variability may explain why different clinicians have had the strong opinions that no “pathognomonic” disk appearance exists.
Kline and Glaser (52) modified the criteria for the diagnosis of DOA as first proposed by Smith. These criteria are listed in Table 22.2. No reports on the treatment of DOA have been published to date, DOA might benefit from idebenone, but further studies are needed (47).
LEBER HEREDITARY OPTIC NEUROPATHY
Leber hereditary optic neuropathy (LHON) rarely has its onset in adulthood, the disease being found predominantly in pediatric- and adolescent-age groups. Although Leber initially described the disease as appearing between the ages of 18 and 23 years, numerous reports now exist documenting its occurrence before age 10 and essentially at any age.
LHON is a maternally inherited disorder and has a 9:1 male-to-female ratio. Approximately 50% of males and 10% of females with the genetic defect develop the optic neuropathy. The majority of cases are associated with point mutations in the mitochondrial genome responsible for complex 1 (NADH:ubiquinone oxidoreductase). These genes are referred to as the ND genes, and the point mutations are as follows: G11778A in ND4, G3460A in ND1, and T14484C in ND6. Testing is important because different mutations have different rates of spontaneous improvement in LHON. Not all LHON patients however have one of these common mutations. Currently, ND6 is considered a hot spot because at least seven different point mutations in this gene have been found in pedigrees with LHON (53,54).
FIGURE 22.7. A and B: Dominant optic atrophy (DOA) demonstrating pallor of the temporal aspect of both disks with focal excavation of the temporal portion of the nerves (courtesy of Dr. Joel S. Glaser).
Presentation
The presenting symptoms of LHON are virtually indistinguishable from those of optic neuritis (ON). Patients report a blur or shadow in their central vision. The onset may be unilateral or bilateral. When one eye is first involved, the second eye invariably becomes affected within several days to weeks; exceptional cases are described with intervals of 3, 8, 12, and 14 years before the second eye became involved.
Table 22.2 DIAGNOSTIC CRITERIA FOR DOMINANT OPTIC ATROPHY (DOA)
Autosomal-dominant inheritance pattern (may benecessary to examine asymptomatic family members)
Insidious onset, often around 10 years of age
Bilateral, symmetric visual loss, although asymmetric loss can occur
Mild-to-moderate reduction in visual acuity
Central and cecocentral scotomata with normal peripheral isopters OU
Tritan dyschromatopsia with possibly superimposed protan and deutan defects; rarely, generalized dyschromatopsia may be present
Temporal disk pallor with possible triangular temporal excavation of disk
Adapted from Kline LB, Glaser JS. Dominant optic atrophy: the clinical profile. Arch Ophthalmol 1979;97:1680-1686; and from Smith DP. The assessment of acquired dyschromatopsia and clinical investigation of the acquired tritan defect in dominantly inherited juvenile atrophy. Am J Optom 1972;49:574-588.
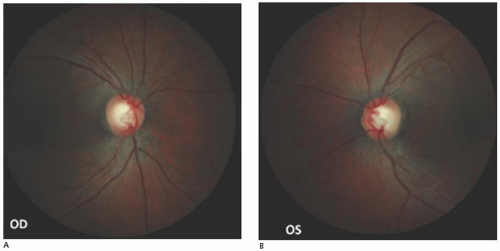
Smith and colleagues (55) postulated that a typical fundoscopic picture exists for acute LHON. They described pronounced telangiectatic microangiopathy in the circumpapillary region associated with hyperemia of the disk and swelling of the nerve fiber layer surrounding the disk (Fig. 22.8). Fluorescein angiography reveals that the peripapillary disk vasculature is dilated but does not leak dye. The disks eventually become pale, with subsequent loss of the retinal nerve fiber layers.
Most patients with LHON have only optic nerve disease, but there are well-documented cases of more diffuse neurologic involvement (56). However, no specific pattern of involvement elsewhere in the CNS has yet emerged. Two patients with electrocardiographic abnormalities have also been described (57), but again this seems more the exception than the rule.
Treatment
A small percentage of patients with LHON demonstrate spontaneous visual improvement. To date, it is impossible to predict who will regain visual function and to what degree. At the present time, no therapeutic regimen has been documented to be effective.
Pfeffer et al. reviewed eight which showed no clear evidence supporting the use of any intervention in mitochondrial disorders. Further research is needed to establish the role of a wide range of therapeutic approaches (47,58). General therapies for mitochondrial diseases have been proposed, including vitamins and cofactors (coenzyme Q10 [CoQ10], folic acid, vitamin B12, thiamine, riboflavin, L-carnitine, L-arginine, and creatine); electron acceptors (vitamin C, menadiol); free radical scavengers (CoQ10, idebenone, α-lipoic acid, minocycline, cyclosporine A, glutathione, and vitamin E); and inhibitors of toxic metabolites (dichloroacetate) (59,60).
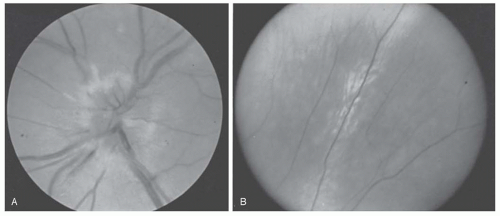
FIGURE 22.8. A and B: Optic atrophy involving papillomacular bundle (temporal pallor) in left and right eyes in Leber hereditary optic neuropathy.
FIGURE 22.9. B: The temporal peripheral retina of the patient shown in (A). The perivascular RPE change and sheathing of this retinal vessel developed 3 months after the acute papillitis.
OPTIC NEURITIS
Optic neuritis (ON) refers to an idiopathic or demyelinating condition of the optic nerve. In contrast with the adult form of this disease, ON in children has three differentiating characteristics: (a) bilateral simultaneous involvement is much more common in children than in adults, (b) between 70% and 80% of children demonstrated optic disk edema compared with approximately 30% of adults with this disorder (Fig. 22.9), and (c) association with multiple sclerosis (MS) is much lower in children than in adults. It is unclear whether this difference related that environmental or genetic risk factors in children compared with adults. There are no published studies comparing incidence rates or prevalence ratios from population-based multiethnic cohorts in either children or adults with MS or other forms of acquired demyelinating syndromes (ADS) (61).
The incidence of acquired demyelination of the CNS (ADS) in children is unknown. The annual incidence of pediatric ADS was between 0.3/100,000 and 1.66/100,000. ON was one of the most common subtypes of ADS accounting for 23% to 36% with an estimated prevalence of 3.2/100,000 (62,63,64).
ON in children is most often associated with postinfectious etiology with symptoms of febrile or flu-like illness preceding the ON by days to weeks. In addition, infectious, systemic inflammatory, and postimmunization causes are all possible (65,66,67).
In the pediatric population, an accurate history of visual loss is often difficult to determine and the vision loss frequently bilateral (33% to 86%) and profound (<20/200 in 90% to 95% of children) (62,68).
Visual improvement starts within 3 weeks of the visual loss, and continues up to 6 months. Younger children (<6 years of age) have a better visual prognosis than older children. Optic disk appearance was abnormal in 83.3% of the eyes in the acute phase. At presentation, RAPD was detected in 67%, visual field defects in 58.5%, and color vision defects in 50% of eyes (69).
Visual evoked potentials (VEP) are of value in confirming clinical suspicion of ON, VEP signal is initially absent in eyes with acute ON, latency remains abnormal in 45% to 65% of children up to 6 to 12 months (62,70).
If ON is suspected clinically, then neuroimaging should be performed to rule out demyelination, inflammatory disease, or less likely an intracranial mass or evidence of hydrocephalus. Underlying infectious causes, as well as increased intracranial pressure (ICP), are ruled out by performing a lumbar puncture. Pediatric ON conversion to MS ranges from 0% to 33%. Bilateral sequential or recurrent ON was associated with a higher rate of MS conversion (62,71,72). A positive MRI scan for demyelinating white matter lesions at ON onset is a strong predictor for development of MS (68,73).
Treatment of ON in the pediatric population remains controversial. To date, there has been no prospective randomized trial to determine the role of systemic corticosteroids in the treatment of pediatric ON. On the basis of the adult Optic Neuritis Treatment Trial (74,75), which showed faster visual recovery with high-dose intravenous steroids followed by oral steroids, ON is often treated with steroids (76).
DEVIC DISEASE
Devic disease (neuromyelitis optica [NMO]) is a variety of CNS demyelinating disease confined as severe acute transverse myelitis (TM), ON, or both. Recurrent NMO is 3 to 9 times more prevalent in women than in men, while in the monophasic form the female/male gender ratio is 1:1. NMO can be a fulminant monophasic or a polyphasic disease with multiple relapses and clinical remissions and with varying degrees of recovery (77,78). NMO-IgG, firstly identified by indirect immunofluorescence in patients with opticospinal variant of CNS demyelinating disorders, is an autoantibody of an IgG isotype specific for Aquaporin4 (AQP4). AQP4 is the most abundant water-channel protein in the CNS (79).
In adults, the sensitivity of NMO-IgG is 73% and specificity of 91%, and in children sensitivity ranges from 67% to 80%.
The high specificity of NMO-IgG for NMO, is coincident with clinical relapse. Children and adults may be affected with this disorder, which produces bilateral visual loss and TM. The visual loss is always bilateral, but it may begin in one eye and involve the second eye several days later (80). Paraplegia from TM quickly follows. In addition to white matter lesions, the gray matter of the spinal cord may be involved. Compared with those who have MS, patients with Devic disease demonstrate a much more elevated cerebrospinal fluid (CSF) protein concentration, as well as increased numbers of inflammatory cells in the spinal fluid. The treatment outcome of NMO is variable, unpredictable, and in many cases unsuccessful. No specific protocols are there regarding the optimum treatment regimen. IV corticosteroids are commonly used for an acute attack of ON or TM. For resistant cases, plasmapheresis, immunosuppressive therapy with (azathioprine, mitoxantrone) IV immunoglobulin, mycophenolate mofetil, and rituximab are recommended to reduce the risk of relapses (81,82,83,84,85,86).
Only gold members can continue reading. Log In or Register to continue