Pediatric Neuro-Ophthalmology: General Considerations and Congenital Motor and Sensory Anomalies
R. Michael Siatkowski
Joel S. Glaser
This faculty of searching for the object is slowly acquired in the child: and, in truth, the motions of the eye are made perfect, like those of the hand, in slow degrees. In both organs there is a compound operation: the impression on the nerve of sense is accompanied with an effort of the will, to accommodate the muscular action to it.
The Hand, Its Mechanism and Vital Endowments
Sir Charles Bell, 1840
In Bell’s early treatise on the function of the hand, he suggests that both the hand and the eye perfect their motor function through practice, meaning the brain teaches itself to coordinate visual and tactile perceptions. Thus, the brain actively orients the retinal receptors (or the hand) toward a target of interest, and then moves them with increasing precision. Perhaps it is not too far-fetched to compare the fine sense of touch at the fingertips, especially the thumb and index finger, to the fine sense of seeing at the macula. For development of effective eye–hand coordination, the head must become a stable platform for the visual system; however, in infants the neck muscles are weak. In an interesting experiment, it was found that, when the head is supported in an upright position, babies of 5 to 8 weeks of age reach with the accuracy of infants 20 weeks old.1 So, the visual system waits for a stronger neck.
The elegant complexity of the developing visual system is a recurring theme throughout the practice of pediatric neuro-ophthalmology, a unique subspecialty because it straddles three medical fields. Ophthalmologists are at the forefront of diagnosing and managing patients with combined visual, neurologic, and systemic abnormalities. Neuro-ophthalmologic problems of congenital origin or childhood onset may come to diagnostic attention in infancy (the blind baby), later in childhood (the grade school child with migraine), or in some cases, not until adulthood, when a congenital motor or sensory defect may be attributed to an acquired abnormality. For example, Duane retraction syndrome may be confused with a sixth nerve palsy, or anisometropic amblyopia may be interpreted as an acquired optic neuropathy. This chapter reviews and summarizes many of the important entities that fall into these potentially confounding categories.
THE PATIENT ENCOUNTER
Children are not little adults and infants are not even little children. Accordingly, the neuro-ophthalmologic history in infancy and childhood must be focused selectively; inquiries often must be directed toward the details of the pregnancy and postpartum period, including the gestational age, maternal illnesses, medications used during pregnancy, infant birth weight, Apgar scores, and use of perinatal oxygen. A detailed family history of ocular, neurologic, or musculoskeletal problems, as well as parental consanguinity must be explored. Neurodevelopmental milestones (Table 1) should be documented when indicated; height, weight, and head circumference should be charted on standard nomograms.
Table 1. Neurodevelopmental Milestones | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Because the attention span of infants and children is quite limited, the physical examination must be conducted in a relatively brief time period. Simple inspection may allow the examiner to assess skull, facial, and other physical anomalies; note the presence of lid malposition or globe displacement; evaluate spontaneous ductal eye movements or the presence of nystagmus; and assess cognitive and to some extent, visual function.
Vision in infants is evaluated by noting response to light or hand–motion threat, the accuracy of the fixation reflex toward a hand light or other attractive object, and the capacity of each eye to follow during contralateral occlusion. In many cases, an infant’s gross objection to occlusion of one eye, by crying or avoiding or pushing away the occluder, provides the first clue that vision in the uncovered eye is significantly impaired. Elicitation of optokinetic nystagmus assesses a visual sensory, as well as a motor, response.
The measurement of grating acuity by forced preferential looking tests (Teller acuity cards) is arguably the most useful method of obtaining reliable, reproducible, quantifiable information on visual acuity in infants and nonverbal children (Fig. 1).2,3,4 However, some have questioned the definition of “normal limits.” Kushner et al5 noted the often poor correlation between Teller (grating) acuity and Snellen (optotype recognition) acuity, the former yielding significantly better results, particularly when Snellen acuity is less than 20/70. It has been suggested that Teller acuity card testing results are imprecise and should not be used by social service agencies to determine legal blindness.6 Vernier acuity also may be measured in infants and children using preferential looking techniques.7 This may be more sensitive than grating acuity in detecting visual changes resulting from amblyopia. Nevertheless, grating acuity measurement is a practical method to assess visual function in the outpatient office setting; it is a valuable asset for determining whether vision is in a normal range for age, and it is useful for following progression of disease or visual development. Various clinical strategies for measuring visual acuity in children are summarized in Table 2.
Table 2. Clinical Applications of Vision Testing in Children | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
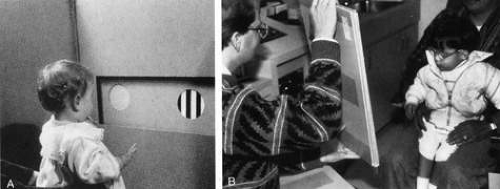 Fig. 1. A. The examiner, masked to orientation of the stripes in the cards, evaluates visually directed eye movements during Teller acuity card testing. B. Cards may be presented at closer distances for children with low vision; vertical presentation is particularly helpful for patients with horizontal nystagmus. |
Infants and children present particular challenges for successful visual evoked potential (VEP) recording, including their sleep–wake status, often poor postural positioning of the patient, and the inability to control and monitor ocular fixation and accommodation. Overestimation of acuity by VEP, particularly in amblyopia, may occur8; thus, for each laboratory, normative data for age group must be established. In many centers, VEP testing for the purpose of measuring acuity is unrewarding and inaccurate. However, in standardized laboratories with experienced personnel who are comfortable and familiar with administering the test, the visual evoked response, particularly pattern VEP, may be useful for monitoring visual development in both preterm and full-term infants.9
The visual field in infants may be assessed by gross confrontation mechanisms, such as visually evoked eye movements or withdrawal or fright to threatening maneuvers (see Chapter 2, Figs. 13,14,15). Hemifield defects are easily detected when a child regularly fails to look at an object of interest (e.g., a toy) held in the nonseeing field. The patient’s response to simultaneous presentation of objects in both hemifields can be assessed. Visually elicited eye movements are noted quite easily by the examiner. For more quantitative visual field measurements in infants, a variant of arc perimetry can be used. The work of Mohn et alsupplies useful normative data regarding normal development of binocular and monocular visual fields during the first year of life.10 By 3 years of age, children are able to comply with finger-mimicking tests. An experienced examiner frequently is able to perform simple kinetic perimetry in children older than 5 years, and many children aged 7 years and older are able to perform suprathreshold automated static perimetric testing, which may be presented as a “video game” with rewards for “good scores.”
The American Academy of Pediatrics has established guidelines for visual assessment.11 Children from birth through 3 years of age should have an eye evaluation in the physician’s office to include: ocular history, vision assessment, external inspection of eyes and lids, ocular motility assessment, pupil examination, and red reflex examination. After age 3 years, evaluations should include all of the former, as well as more age-appropriate visual acuity assessments and an attempt at ophthalmoscopy. Appropriate testing strategies and referral criteria for distance acuity, ocular alignment, and assessment of ocular media clarity are detailed in the preceding reference.
VISUAL DEVELOPMENT
Visual and ocular developmental milestones are summarized in Table 3. The “critical period” of visual development refers to the period early in infancy when the visual system is sensitive to deprivation. It is believed that two different types of visual processing occur. Magnocellular (M) pathways deal with perception of motion, whereas parvocellular (P) neurons are more attuned to image shape and color (see Chapter 4). These two pathways are separated at the retinal ganglion cell level through to the lateral geniculate nucleus, and are represented in different areas in layer V-1 of the striate cortex. The parieto-occipital extrastriate cortex deals more with motion perception, whereas the temporo-occipital region is concerned with image shape and color. There is direct neural communication between these two areas. Maldevelopment of the magnocellular system occurs with infantile esotropia, latent and congenital nystagmus, impairment of gross stereopsis, and motion VEP deficits. Maldevelopment of the P system is associated with anisometropic and strabismic amblyopia, deficits in fine stereopsis, and spatial sweep VEP deficits.12
Table 3. Visual and Ocular Developmental Milestones | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Although normal binocular vision requires aligned eyes with intact sensory and motor fusion mechanisms, in the first few months of life some children have intermittent or constant ocular deviations, most commonly exotropia. Nevertheless, as the strength of the neck muscles permits (see earlier) in normal infants at about 3 months of age, there is a relatively rapid maturation of binocularity, including fusional convergence and sensory fusion (stereopsis). It has been hypothesized that refinement of cortical circuits, particularly those in the ocular dominance columns, occurs during this period of instability.13 It is well documented that children with esotropia, amblyopia, or early visual deprivation have asymmetric monocular pursuit noted on optokinetic nystagmus testing, favoring targets moving in a temporal-to-nasal direction; however, the presence of normal stereoacuity and binocular function is not always sufficient to prevent this bias.14 The relationship among binocularity, fusion, visual deprivation, and monocular optokinetic nystagmus asymmetry is complex and not precisely understood.
Visual development in infants with severe ocular disorders (e.g., retinal dystrophies, congenital optic nerve anomalies) may show delayed improvement of assessable visual function later in childhood, even up to the end of the first decade of life.15 Continuing posterior visual pathway maturation may be the mechanism responsible for late visual improvement in some cases. Of course, significant improvement in visual acuity as a result of amblyopia therapy also may occur in patients with either unilateral or bilateral, asymmetric structural abnormalities.16 It is imperative that the clinician exercise restraint when attempting to predict final levels of visual function in such children. In clinical practice frequently it is not possible to estimate visual function from either fundus appearance or neuroradiographic status of the anterior and posterior visual pathways.
THE BLIND INFANT
Assessment of the infant with subnormal or absent visual responses usually distinguishes between two major groups: those with and those without congenital (idiopathic infantile) nystagmus. Congenital nystagmus (see the following) may appear as a jerk or pendular, horizontal, uniplanar eye movement disorder, which frequently diminishes on convergence and is absent during sleep. In the past, congenital nystagmus has been divided into “sensory” and “motor” types. Although these concepts may be helpful clinically, they do not accurately reflect the underlying pathophysiology or the complex foveation strategies that the ocular motor system employs. It should be noted that a variety of other clinical features may be encountered in congenital nystagmus, including inversion of the optokinetic response and vertical or torsional movements (see Chapter 11).
Congenital nystagmus occurs in 1 in 1,000 to 6,550 births, and it is frequently associated with bilateral visual loss of prechiasmal or chiasmal origin.17 When occurring in the presence of bilateral corneal or lens opacities, optic nerve disorders, or macular disease, the diagnosis is straightforward. In the infant population, Gelbart and Hoyt18 demonstrated anterior visual pathway disease in 119 of 152 patients with congenital nystagmus, including diagnoses of bilateral optic nerve hypoplasia, Leber’s congenital amaurosis, and ocular or oculocutaneous albinism. Likewise, Cibis and Fitzgerald19 noted bilateral retinal disease in 56% of patients with congenital nystagmus. Unilateral anomalies, however, allow development of normal foveation in the sound eye and, with rare exception, are not associated with congenital nystagmus. Often, congenital nystagmus does not present until the second to fourth month of life, when central, steady fixation and foveation should be established in normal infants.
In an infant with poor vision and nystagmus, but with an apparently otherwise normal eye examination, electroretinography (ERG) should be performed. Lambert et al20 reviewed this clinical scenario. Leber’s congenital amaurosis is an autosomal-recessive retinal rod/cone disorder that causes poor vision from birth. At least seven different genetic defects have been shown to cause a similar clinical picture, and there is good evidence that photoreceptor degeneration actually starts prenatally.21 Although initial fundus examination may be unremarkable, pigmentary retinopathy, vessel attenuation, and optic atrophy occur over time. ERG shows nonrecordable or grossly attenuated electrical potentials of both rods and cones (“flat ERG”) (Fig. 2). Although initial acuity is quite poor, it is unlikely to deteriorate substantially over the course of the patient’s life.22 No treatment is available. In exceptionally rare cases, spontaneous visual improvement may occur23; pharmacologic treatment in a mouse model has also been reported to be effective in improving vision.24
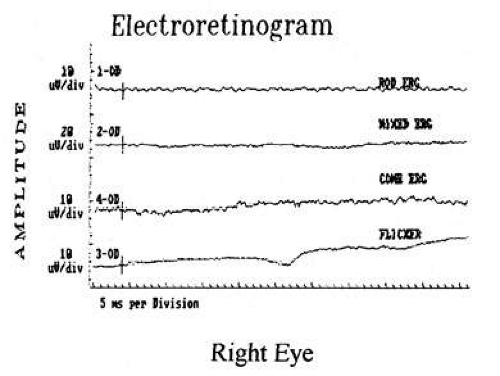 Fig. 2. Electroretinography from patient with Leber’s congenital amaurosis demonstrates absent rod and cone responses. |
Achromatopsia (rod monochromatism) is an autosomal-recessive or X-linked disease characterized by a partial or complete absence of retinal cone function. Acuity generally is better than in Leber’s amaurosis and frequently exceeds 20/200. In this condition, the rod ERG is normal, whereas the cone ERG is significantly impaired (Fig. 3). Photophobia, which may be marked, is quite common, and these children show marked preference for dim lighting. Other hereditary retinal disorders may be seen in Joubert’s syndrome (cerebellar vermis hypoplasia), congenital stationary night-blindness, Refsum’s disease, neonatal adrenoleukodystrophy, neuronal ceroid lipofuscinosis, Jeune’s syndrome, and osteopetrosis.
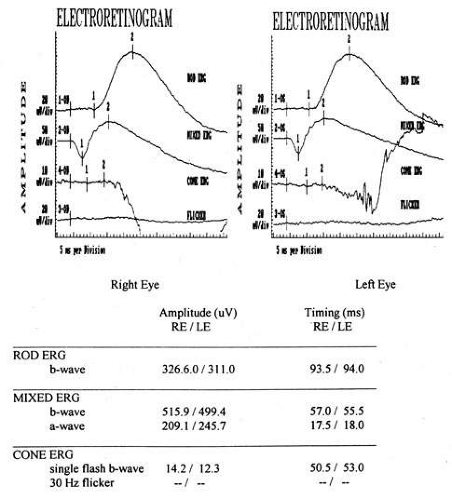 Fig. 3. Electroretinography from a patient with achromatopsia. Rod (top) and mixed (second) tracings are normal, but the cone flash and flicker responses (bottom two tracings) are severely reduced or absent. |
Patients with albinism (Fig. 4) and aniridia, both of which may be associated with foveal (and optic nerve) hypoplasia, may present with congenital nystagmus. Anomalous (excessive) chiasmal crossing defects and cortical miswiring25 have been reported in ocular albinism. VEP in albinism demonstrates that the majority of visual fibers decussate at the chiasm, rather than about half as in normal individuals.
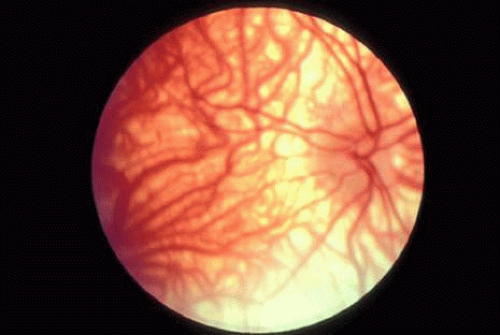 Fig. 4. Fundus of patient with albinism. Note marked absent of melanin in the RPE, with prominent choroidal vessels and foveal hypoplasia with lack of foveal reflex. |
Note that the nystagmus in association with disorders of bilateral anterior visual pathway disease should not be confused with the bilateral “manifest latent nystagmus” seen in patients with monocular decreased vision, which acts as an occluder, thus manifesting what otherwise would have been truly latent nystagmus (see Chapter 11).26
Optic nerve hypoplasia, unless subtle, usually is diagnosed on fundus examination. Although most cases are idiopathic, maternal gestational diabetes and use of phenytoin are well-known risk factors; more recent epidemiologic studies have also suggested that young maternal age, first parity, smoking, and use of fertility and antidepressant drugs may also play a role.27 If bilateral, this condition may be associated with congenital nystagmus. De Morsier’s syndrome (septo-optic dysplasia) refers to the constellation of bilateral optic nerve hypoplasia, absence of the septum pellucidum (Fig. 5), thinning or absence of the corpus callosum, dysplasia of the anterior third ventricle, and pituitary dysfunction (see Chapter 5, Fig. 9). Brodsky and Glasier28 broadened the spectrum of this condition. In a study of 40 children, some optic nerve anomalies were isolated, but in other children, midline craniofacial defects, hemispheric gray matter dystrophic anomalies, and posterior pituitary ectopia were noted. Of 21 cases of optic nerve hypoplasia described by Zeki et al,29 there were midline central nervous system defects in six and endocrine deficiencies in nine. In the series of 35 patients with bilateral optic nerve hypoplasia described by Siatkowski et alet al30 neuroradiographic abnormalities were seen in 46% and endocrinopathies in 27%. Growth hormone deficiency was the most common endocrine abnormality. The visual spectrum ranged from 20/20 in one case to no light perception in 34% of patients; 80% were legally blind (20/200 or less in both eyes). Absence of the septum pellucidum and corpus callosum, with panhypopituitarism, occurred in only 11.5% of all patients with bilateral optic nerve hypoplasia. Recently, a sporadic mutation in the HESX1 gene has been reported to cause optic nerve hypoplasia with pituitary insufficiency.31
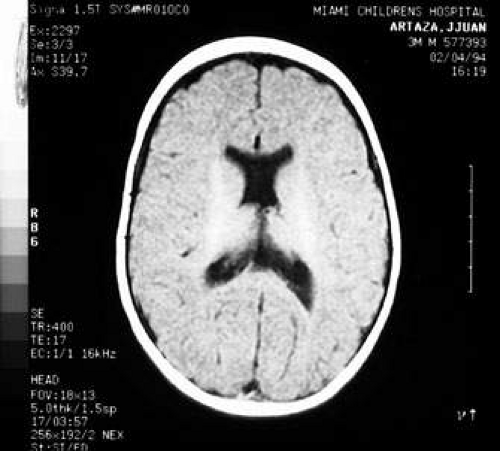 Fig. 5. Magnetic resonance image of patient with bilateral optic nerve hypoplasia demonstrating absence of septum pellucidum with single midline ventricle. |
A variant of this condition has been termed “superior segmental optic nerve hypoplasia,” wherein central acuity may be spared, but affected patients have inferior altitudinal field defects and superior optic pallor with nerve fiber layer dropout; maternal diabetes may be an associated factor.32 Additionally, it has been proposed that mild cases of optic nerve hypoplasia may indeed be misdiagnosed as amblyopia. Lempert demonstrated a significant difference in mean disc area and axial length in presumed amblyopic eyes compared to non-amblyopic controls.33
Optic disc colobomata (Fig. 6), if bilateral, may be seen in association with congenital nystagmus. Of greater import, however, is the association of colobomas or other dysplastic optic discs with basal encephaloceles, particularly in conjunction with hypertelorism, midfacial anomalies, or tongue-shaped retinochoroidal pigmentary disturbances.34 The morning glory disc syndrome, although typically unilateral, does not present in association with nystagmus, but rarely may be seen with midline central nervous system defects and endocrinopathies as well.35
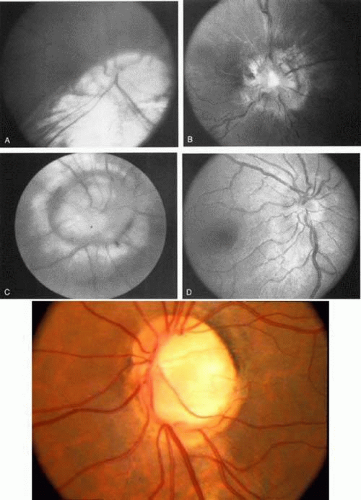 Fig. 6. Spectrum of congenital disc anomalies. A. Large coloboma involving optic nerve and inferior retina. B. Dysplastic disc, central cup filled with fibroglial tissue. C. Morning glory disc anomaly. D. Markedly hypoplastic optic nerve. E. Coloboma of optic nerve without retinal involvement. |
The infant with poor vision, no nystagmus, and a normal eye exam is likely to have cortical visual impairment. Good et al36 extensively reviewed this entity. These patients have normal pupillary light reaction, unremarkable fundi (at least initially), and absence of nystagmus in all but the rarest cases; however, they do have various neurobehavioral signs of cortical visual impairment:
They have a preference for brightly colored objects and stare at bright lights (although up to one-third may have photophobia).
They adopt peculiar head turns when attempting to look at or reach for an object of interest, because peripheral visual field, rather than central vision, is used.
They may use extrageniculate vision, or “blindsight,” in some cases enabling them to localize targets or to see colors. This concept remains much in debate however, and many instances of “blindsight” may, in fact, result from preservation of some portions of visual cortex.
They exhibit varying degrees of alertness and employ idiosyncratic visual “strategies” to use patches of visual field remnants.
Prognosis for recovery depends on etiology, age of onset, and severity of brain damage (Fig. 7). Although such children often remain visually handicapped, dramatic recovery may ensue occasionally.36 Cortical visual loss from perinatal hypoxia/ischemia has a particularly poor prognosis if congenital, but up to a 70% recovery rate if acquired.37 Although hydrocephalus, brain dysgenesis, and infections may produce cortical visual loss, by far the most common cause of CVI in the developed world is perinatal hypoxia. In term infants, damage primarily affects the watershed areas of the cerebral cortex (frontal and parieto-occipital regions). In premature babies, however, the damage is predominantly periventricular. Age-related differences in the developing intracranial vascular systems may account for the disparate sites of injury. The watershed zone in term infants (and adults) is at the parieto-occipital junction, whereas in preterm infants it is in the subcortical area.38
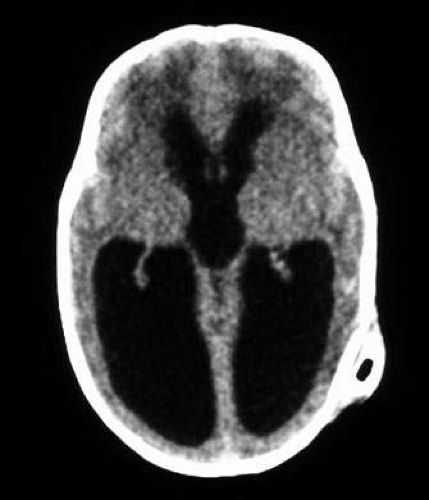 Fig. 7. Computed tomography of child with cortical visual impairment resulting from congenital hydrocephalus. Note significant ex vacuo dilatation of the ventricles posteriorly, with shunt catheter in place. |
Although initially unremarkable in appearance, the optic disc subsequently may become abnormal in some cases of cortical visual impairment. Periventricular leukomalacia (PVL), which selectively involves the optic radiations near the trigone of the lateral ventricle, as well as the anterior corticospinal fibers, may produce abnormalities of visual acuity and visual field, disorders of higher cortical visual function, as well as spastic diplegia. Likely resulting from trans-synaptic degeneration across the lateral geniculate nucleus, PVL may produce secondary optic disc cupping, which may be confused with normal-tension glaucoma.39,40 As PVL primarily involves the retrogeniculate white matter, Brodsky has suggested that it be reclassified as “subcortical visual loss,” noting that such patients may have optic disc pallor and apparent hypoplasia in addition to cupping. In contrast, preferential involvement of the gray matter in the visual cortex carries with it a much lower incidence of optic disc abnormalities. Children with subcortical visual loss (PVL) also are more likely to exhibit tonic downgaze (sunsetting) and esotropia, whereas those with gray matter (cortical) disease have a higher incidence of exotropia and dysconjugate horizontal gaze.39,40 As PVL primarily often are present early in infancy, optic atrophy and cupping may not become apparent until later in the first, or seven second, year of life, as transsynaptic degeneration ensues.
Delayed maturation of vision is a diagnosis of exclusion and retrospection.41 This pertains to visually impaired infants with normal ocular examinations, normal neuroimaging and ERG, and no nystagmus. The exact etiology is unknown. Delayed myelination of the posterior visual pathways has been implicated, but in most cases, magnetic resonance imaging (MRI) shows age-appropriate myelination. Another hypothesis is that eye movement abnormalities (apraxia) may be misinterpreted as poor vision, as assessment of visual behavior in infants is to large degree interpreted based on visually directed ocular movements. Thus, care should be taken in making a diagnosis of delayed visual maturation in patients with abnormal saccades. More recent psychophysical studies suggest that delayed development of the subcortical (extra-geniculostriate) pathways may be the primary site of the defect.42,43,44 Alternatively, Hoyt has suggested that there is no evidence for failure of any primary visual system, and that a temporary visual inattention is the cause of this problem.44A
In this entity, visual function spontaneously improves, often quite rapidly over several days, suggesting a discrete rather than a widespread structural abnormality. This retrospective diagnosis should be made only when visual function indeed spontaneously improves to normal or near-normal levels, as best ascertainable by the end of the first year of life. However, careful visual testing later in childhood often discloses less than 20/20 acuity and various levels of binocular dysfunction; neurologic milestones may be delayed and cognitive function often is below average.
Children with unilateral hemianopic defects may adopt a head turn toward the side of the field defect so that the eyes are rotated toward the objects of visual interest. In addition, many of these children develop an exotropia,45,46 possibly to increase the panoramic visual field in compensation for the underlying hemianopic defect. It has been suggested that strabismus surgery in these children, therefore, should be avoided.
ACQUIRED VISUAL LOSS IN CHILDHOOD
Hereditary optic atrophies, neuroretinitis, optic nerve tumors, and trauma are discussed in Chapter 5 of this text. However, a few statements are in order regarding several entities.
Optic neuritis has notably different characteristics in the pediatric population than in adults. Various authors have reviewed this entity.47,48,49,50,51,52 Childhood optic neuritis, particularly in prepubescents, commonly presents as simultaneous bilateral papillitis (see Fig. 8, also see Chapter 5, Fig. 22); unlike adults, there is no clear sex predilection in this age group. Antecedent vaccinations or viral illnesses, ranging from upper respiratory infections to varicella, mumps, and mononucleosis, may precede visual loss by 2 to 6 weeks; some clinicians regard pediatric optic neuritis as a forme fruste of acute disseminated encephalomyelitis (ADEM). Initial visual acuity is typically poor, 20/200 or less, in many instances. Cerebrospinal fluid pleocytosis and elevated protein are common, but the eventual association with multiple sclerosis is less frequent in children than in adults.53 Prompt responses (within days) to systemic corticosteroids in doses ranging from 1 mg/ kg prednisone to high-dose intravenous methylprednisolone are the rule. Steroid taper over several weeks is recommended to reduce rebound inflammation. Although good visual outcomes are achieved in the majority (50% to 75%) of cases, there appears to be a higher rate of permanent visual deficits than in the adult population. Although MRI may demonstrate optic nerve enhancement or cortical white matter lesions in some cases, the results of the Optic Neuritis Treatment Trial (see Chapter 5) are not applicable to children. Later in childhood and in adolescence, optic neuritis begins to take on more of the characteristics seen in adults (i.e., female predilection, unilateral disease, retrobulbar neuritis, final visual acuity greater than 20/40 in over 90% of cases, and increasing likelihood of developing multiple sclerosis).52
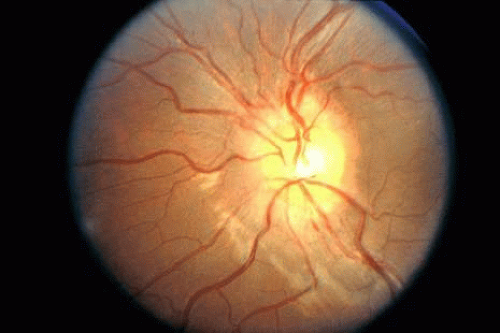 Fig. 8. Fundus photo of 10-year-old girl with bilateral optic neuritis demonstrates mild optic nerve edema without hemorrhages or exudates. The fellow eye appeared similar. Vision was 20/400 in each eye. |
Leber hereditary optic neuropathy (Fig. 9) commonly presents in the second decade of life,35 but several cases occurring within the first decade also have been reported.54,55,56,57 There is some evidence that those patients with the mtDNA 14484 mutation presenting in the second decade may show a higher incidence of spontaneous visual recovery, generally within 12 to 18 months of the initial visual loss. Because the initial fundus appearance in such patients often is unremarkable (especially if the typical telangiectatic peripapillary vessels are not present and there is no relative afferent pupillary defect in bilateral disease), children with Leber hereditary optic neuropathy may erroneously be misdiagnosed as functional visual loss at first presentation. Serial examinations are appropriate in such cases. In addition to Leber type, both dominant and recessive hereditary optic neuropathies may present in the first or second decades of life (see Chapter 5).
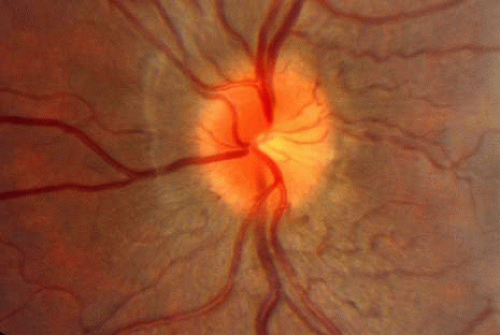 Fig. 9. Sixteen-year-old boy with Leber hereditary optic neuropathy. Vision was 20/400 with large central scotoma. Note minimal disc swelling and peripapillary telangiectatic vessels. The fellow eye became involved 1 year later. |
Patients with both dominant and recessive hereditary optic neuropathies may complain of a slow deterioration of vision throughout the first and second decades of life. Dominant disease rarely causes acuities less than 20/200, but in recessive disease it may be 20/400 or worse. Another hereditary condition with optic neuropathy is Wolfram syndrome, also called “DIDMOAD,” for diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (Fig. 10). Further information on hereditary optic neuropathies is found in Chapter 5.
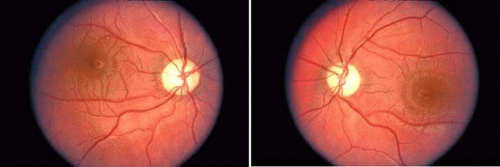 Fig. 10. Six-year-old boy with Wolfram (DIDMOAD) syndrome. Note symmetric temporal pallor of both optic discs. Vision was 20/60 in each eye. |
Anterior visual pathway gliomas account for the majority of intrinsic optic nerve tumors in childhood (Figs. 11, 12). Although they are true neoplasms, malignant features are extraordinarily rare in the pediatric population (see Chapter 5). Dutton58 provided a thorough review of this subject. When the glioma is initially confined to the optic nerve alone, the mortality rate is 5%. However, when the hypothalamus is involved, survival is less than 50% in some series. With a typically indolent course, these tumors generally can be managed conservatively, especially when confined to the optic nerve. Hoffman et al59 reviewed 62 cases of optic pathway/hypothalamic gliomas over a 14-year period, with 48 of these exhibiting relative stability with only visual defects: six patients had significant neurologic abnormalities, and eight died. Gayre et al60 reported a series of 42 patients with optic gliomas seen over 28 years at a single institution. Two-thirds were female, and slightly over half had neurofibromatosis type I (NF-1). Presenting signs and chiasmal involvement were similar in both NF (+) and NF (–) groups, the latter usually occurring within the first year after diagnosis. Regardless of treatment, the eye with better vision tended to remain stable over the long-term (and vision in the poorer eye often declined). Spontaneous regression of optic gliomas with visual improvement also has been reported.61,62
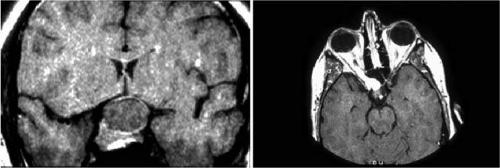 Fig. 11. Magnetic resonance imaging scans of optic gliomata. A. Marked enlargement of optic chiasm on coronal image. B. T1-weighted, gadolinium-enhanced image showing enlargement and marked enhancement of the right optic nerve and chiasm. |
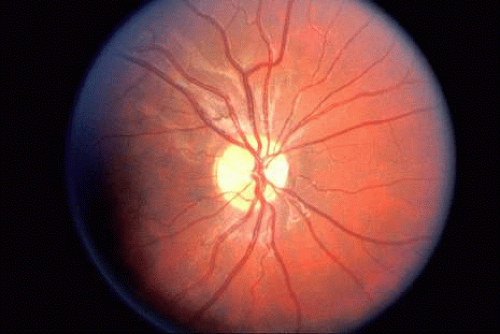 Fig. 12. “Bow-tie” atrophy of the optic nerve in a patient with NF-1 and diffuse optic glioma involving the optic tracts (see Chapter 5). |
Tumors abutting the visual system that may cause progressive visual loss by extrinsic compression, including craniopharyngiomas, are discussed in Chapter 5. Pituitary tumors in childhood, although reported, are extremely rare.
Other causes of progressive visual loss in childhood are those syndromes associated with retinal pigmentary abiotrophies (tapetoretinal degenerations). These have a later onset than the congenital photoreceptor dystrophies, which profoundly affect vision at birth, and generally are stable. Included among these progressive retinal degenerations are the Bardet-Biedl syndrome (an autosomal-recessive disorder of polydactyly, obesity, hypogenitalism, renal disease, and tapetoretinal degeneration); Refsum’s disease; Bassen-Kornzweig’s syndrome (abetalipoproteinemia with intestinal malabsorption); and Kearns-Sayre’s syndrome (progressive external ophthalmoplegia [see Chapter 12]).
Aicardi’s syndrome consists of agenesis of the corpus callosum, chorioretinal lacunae (Fig. 13), and infantile spasms with seizures. The condition is limited to females and, with one exception, is lethal in males. Additional ocular features may include microphthalmia, optic nerve colobomas, peripapillary glial tissue, and variants of persistent fetal vasculature.63,64 Affected children rarely achieve developmental milestones beyond 12 months, and vision is decreased because of combined anterior and posterior visual pathway disease.65
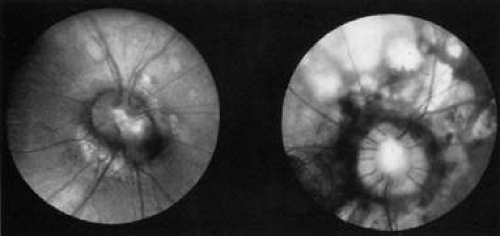 Fig. 13. Fundus of a 3-year-old girl with Aicardi’s syndrome. Note dysplastic optic discs surrounded by hypopigmented retinal lacunae. (Courtesy of John T. Flynn, MD) |
The neurometabolic syndromes may present with progressive visual loss in infancy and childhood. These include, but are not limited to, Tay-Sachs’, Niemann-Pick’s, and neuronal ceroid lipofuscinosis (Batten’s disease). The majority of these conditions also produce various other neurologic symptoms, with developmental regression in virtually all. Visual loss may be on the basis of optic atrophy, retinal degeneration, cortical factors, or a combination of both (see Chapter 5).
The most commonly encountered neurometabolic disease in pediatric neuro-ophthalmology is the group of neuronal ceroid lipofuscinoses (NCLs). These are autosomal recessive conditions characterized by brain and retinal atrophy. Infantile NCL produces generalized neurologic deterioration before visual problems occur, but in juvenile NCL (Batten’s disease), visual symptoms are frequently the first sign of the disorder. Acuity declines to hand motion or worse in half of cases before other problems manifest. Fundus examination reveals optic atrophy and maculopathy, which may range from a subtle grainy appearance to the typical bull’s-eye maculopathy. ERG shows depressed a and b wave responses under both photopic and scotopic conditions. Although many of the gene products responsible for this disorder have been identified in the laboratory, the exact mechanism of disease remains elusive, and treatment is unavailable.66,67
For further information on NCL and other metabolic disorders, the reader is referred to Brodsky et al
CONGENITAL MOTOR ANOMALIES
ANOMALIES OF INNERVATION AND ORBITAL MECHANICS
For many decades, clinical descriptions of eye movements, coupled with gross observations made during surgery, have generated a preoccupation with malformations of extraocular muscles, fascial connections, tendon sheaths, or check ligaments. In addition, the phenomena of “overacting” and “underacting” single ocular muscles are invoked by way of “explanations” of defective motor patterns. Electromyography (EMG) provides a technique whereby the electrical activity of extraocular muscles may be sampled. More recently, special orbital MR imaging techniques under both stationary and dynamic conditions have allowed greater understanding of orbital anatomy and extraocular muscle pulley systems. Thus, an interest in the neural and mechanical bases of paradoxic ocular motor defects has been renewed.
Hoyt and Nachtigaller68 reviewed the evidence for peripheral anomalies in the number and distribution of human ocular motor nerves, and have catalogued examples of omission, substitution, and duplication. In the dissection room, absence of the abducens nerve(s) has been documented, as well as agenesis of the sixth nerve nucleus, despite the presence of normal lateral rectus muscles. The lateral rectus indeed may be aggrandized by the oculomotor nerve in the absence of an abducens nerve, or it may receive a dual innervation from both the abducens and oculomotor nerve. In none of the foregoing anatomic studies was clinical information available, and the incidence of such anomalies of innervation cannot be assessed. Phillips et al69 reported pathologic correlation in a 5-year-old girl with bilateral abducens palsy, who died during surgical correction of convergent strabismus; neither abducens nerve was present, and the corresponding nuclei were diminutive. Rarely is such clinicoanatomic correlation possible, but sufficient material has accrued to validate the concept of “congenital miswiring,” including both failure of innervation and anomalous annexation of extraocular muscles by inappropriate nerve branches.
Simultaneous electrical activity in agonist–antagonist pairs of extraocular muscles (cofiring, cocontraction) has been documented on EMG, further substantiating the role of anomalous innervation in the production of paradoxic eye movements. For example, since the early work of Blodi,70 such paradoxic innervation of one of the horizontal rectus muscles is the most outstanding and consistent feature of Duane’s retraction syndrome.
The outstanding work of Demer et al71,72,73,74,75,76,77 has used dynamic orbital MRI and elegant orbital histoanatomy studies to reformulate many concepts of ocular motor physiology. Each of the six extraocular muscles consists of global and orbital layers, the former contiguous with the tendon and inserting on the globe, the latter inserting on an intraorbital connective tissue ring, forming the muscle pulley. These pulleys guide the path of the muscles and serve as their functional origin. It is these pulleys that shift position during vergence eye movements to stimulate stereopsis, and account for the viability of Listing’s law. The global and orbital layers have different innervations, fiber types, and cellular metabolism. Ectopic or unstable muscle pulleys may result in various types of incomitant strabismus, sometimes simulating cranial neuropathies, overacting muscles, or even supranuclear motor defects. Conventional strabismus surgery often is successful in part by its secondary effects on the muscle pulley systems. Pulley position also may be influenced by the placement of retroequatorial sutures to attach the eye muscles to the globe (retroequatorial myopexy).
Duane’s Retraction Syndrome
Clinicians are quite familiar with the most common form of Duane’s retraction syndrome (DRS): limited abduction of one eye, with globe retraction posteriorly into the orbit and narrowing of the lid fissure on adduction. Both autopsy and electrophysiologic studies are in concordance on the etiology of this condition.78 Matteucci79 reported a case of unilateral Duane’s syndrome with a normal medial rectus, but a hypoplastic lateral rectus and hypoplastic abducens nucleus and nerve. In a patient with bilateral Duane’s syndrome, Hotchkiss et al80 found bilateral absence of abducens nuclei and nerves; both lateral recti were partially innervated by branches from the inferior division of the oculomotor nerve and appeared histologically normal in innervated areas, but fibrotic in areas not innervated. Miller et al81 in a Duane patient who had had eye-movement recordings before death, demonstrated that the left abducens nerve was absent and, as in the previous case, the lateral rectus was innervated by branches from the inferior division of the third nerve, with fibrosis in areas not innervated, but normal in innervated areas. These studies provide elegant support for the hypothesis derived from previous EMG studies showing cofiring of the horizontal rectus muscles on attempted abduction (Fig 14).70,78
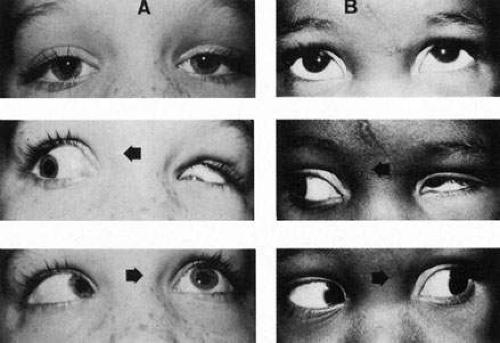 Fig. 14. Duane’s retraction syndrome. A. Type I. Forward gaze, eyes straight with slightly narrowed left fissure. Right gaze, retraction of left globe with downward oblique deviation. Left gaze, failure of abduction. B. Type II. Forward gaze, slight exotropia. Right gaze, retraction of left globe with upward oblique deviation. Left gaze, normal abduction. |
This variant of DRS was labeled type I, with type II described as an isolated adduction deficit, and type III with deficits of both abduction and adduction. However, newer concepts of embryology, coupled with the realization that there are many more twists and turns of aberrant innervation possible, has weakened the traditional classification of DRS.
The review by Mims82 documents that, in the fourth week of embryogenesis, the posterior portion of the abducens nerve in the pons innervates structures of the branchial clefts, which later atrophy as development continues. In most cases of DRS, all or most of the sixth cranial nerve (instead of just the posterior portion) is attracted caudally to these structures. Later in embryogenesis, most or all of the nerve and its nucleus atrophy, leaving the developing fetus either with no sixth nerve or very few residual fibers. The uninnervated (or hypoinnervated) lateral rectus provokes a neurotropic competition among the extraocular muscles in the orbital apex, attracting nerve fibers from the medial rectus subnucleus of the oculomotor nuclear complex. Although this phenomenon is what most likely occurs in the majority of DRS cases, the proximity of these structures during embryogenesis may lead to various other innervational and developmental anomalies, resulting in other motility deficits (e.g., adduction problems, synergistic divergence, abnormal vertical movements), associated synkinetic conditions (e.g., jaw-winking, crocodile tears), cranial nerve dysfunction (e.g., sensorineural deafness), and branchial arch disorders (e.g., Goldenhar’s syndrome). An interesting variant of motility defect has been termed “synergistic divergence,” in which both eyes abduct on attempted lateral gaze.82A
In all types of Duane’s syndrome, vertical deviations of the adducting eye may take the form of upshoots and downshoots (Fig. 15). Scott and Wong83 recorded increased activity in both the inferior oblique and superior rectus muscles during upshoot in adduction, most consistently in the superior rectus, suggesting another variant of aberrant innervation. However, a tightly fibrosed lateral rectus insertion also may act as a mechanical leash, causing dynamic vertical overaction and a so-called flipping mechanism. Meticulous clinical examination and intraoperative assessment of anatomy and forced ductions help to estimate the relative contributions of mechanical and innervational anomalies in DRS-related upshoots and downshoots; then the most effective surgical strategy is possible.84
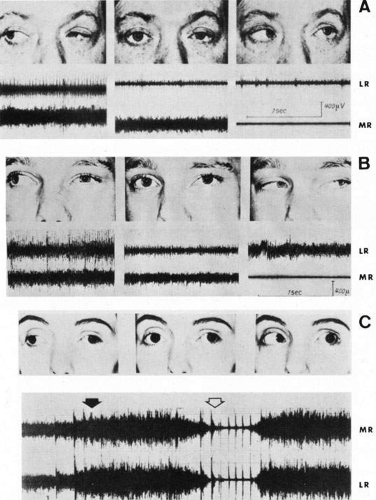 Fig. 15. Electromyography in Duane retraction syndrome. Simultaneous recordings of left lateral rectus (LR) and medial rectus (MR). A. Type I. Peak innervation of lateral rectus on right gaze (paradoxic innervation) and minimal innervation on left gaze. Normal electrical behavior of medial rectus throughout. B. Type II. Lateral rectus innervation on both right and left gaze. Normal electrical behavior of medial rectus throughout. C. Type III. Synchronous and strikingly similar electrical activity in both recti, with intense firing in right gaze (black arrow) and marked inhibition in left gaze (open arrow). (From Huber A: Electrophysiology of the retraction syndromes. Br J Ophthalmol 58:293, 1974) |
When describing individual patients with DRS, five clinical characteristics are important: (a) the type of deviation in primary gaze (esotropic versus exotropic DRS); (b) any compensatory face turn to promote fusion; (c) degree of co-contraction (especially noting globe retraction, adduction deficits, and upshoots and downshoots); (d) type of strabismus present in the reading position (downgaze at near); and (e) degree of abduction deficit. Such details more precisely characterize the condition than the previous nomenclature, and also allow more appropriate planning of corrective surgical strategies.82
According to Raab85 and others, Duane’s syndrome occurs more commonly in women (about 2:1 over men), and the left eye is involved perhaps three times more commonly than the right eye. Approximately 10% to 18% of cases are bilateral. Primary gaze strabismus (usually esotropia) and secondary ocular torticollis are present in approximately 50% of patients. Familial cases are not uncommon, and bilateral retraction has been reported in monozygotic twins.86 Duane syndrome also has been reported in association with abnormalities on chromosomes 4q 87 and 22.88
Patients with DRS may demonstrate a peculiar sensory adaptation, with excellent binocular function in directions of gaze where visual axes are aligned, and suppression without diplopia in the field of the affected lateral rectus (i.e., a facultative suppression scotoma, as is employed in intermittent exotropia). Occasionally, mild amblyopia may be present in the involved eye, but the reported incidence of amblyopia varies greatly, as does the incidence of anisometropia.89 A few patients who as children became accustomed to avoiding the diplopic field may present as adults complaining of an “acute” onset of diplopia. However, microtropia or larger angles of strabismus may be present, convergence often is poor, and sensory adaptations are complex and sometimes tenuous. Age-related deterioration in fusion ability and in convergence, as well as progressive fibrosis of the rectus muscles, can produce symptoms of diplopia and asthenopia. Surgical approaches to the various presentations of DRS are beyond the scope of this chapter, but standard muscle recessions and transposition procedures for primary gaze strabismus and ocular torticollis, and Y-splitting of the lateral rectus for upshoots and downshoots, all may play a role. Surgery on the fellow eye often is required to make ductions and versions more symmetric.
Retraction of the globe somewhat mimicking Duane’s syndrome occasionally may be seen with acquired orbital lesions that cause mechanical restriction. The chronic fibrotic changes of extraocular muscles in thyroid-related orbitopathy may cause a leash effect, most typically observed on abduction attempts, but possible on gaze opposite the field(s) of action of any involved muscle(s). The tethering also prevents full ductional range, and forced-duction testing (see Chapter 12) confirms the restrictive process. Proptosis and other orbital congestive signs generally are present. Similarly, infiltration of muscles by granulomatous inflammation (orbital pseudotumor) or by carcinoma may cause restrictive contraction, but rarely to the degree observed in Duane’s syndrome.90 Medial orbital wall fractures can entrap the medial rectus muscle, with variable abduction limitation and globe retraction,91 and retraction also has been reported with muscle fixations resulting from orbital metastases.92
Möbius’ Syndrome (Congenital Bulbar Palsies)
The discovery in 1888 of the condition of facial diplegia of variable degree associated with paralysis of lateral gaze, and usually esotropia, is attributed to Möbius. Although traditionally considered simply as palsies of the facial and abducens nerves, EMG and neuropathologic evidence indicates a more complex situation, as does involvement of other body systems. Variable associations include tongue hemiatrophy; head and extremity deformities; abnormalities of lower cranial nerves, producing hearing, speech, and swallowing difficulties; chest malformations; congenital heart defects; limb disorders, including club feet; micrognathia and tongue malformations; Poland anomaly; and Kallmann’s syndrome (hypogonadism and anosmia).93,94,95 Clinical studies also have noted a relatively high incidence of mental retardation and cognitive dysfunction96 as well as autism.97 Although generally considered a static neonatal disorder, there have been reports of progressive, increasing motor deficits during later life.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


