ANATOMY AND EMBRYOLOGY
The crystalline lens is a clear, avascular structure suspended behind the iris by the zonules of Zinn. The purpose of the lens is to refract light, accommodate and maintain its own clarity. The lens is comprised of the lens capsule, lens epithelium, cortex, and nucleus.
The lens is derived from surface ectoderm after interacting with the neuroectoderm of the optic vesicle, which occurs at approximately 25 days of gestation. At approximately 27 days of gestation, the surface ectoderm overlying the optic vesicle becomes elongated (columnar) to form the lens plate (also known as the lens placode). By 29 days, the lens pit is formed by indentation of the lens plate. The lens pit continues to invaginate, the stalk of cells connecting the new lens to the surface ectoderm undergoes apoptosis. The resultant structure is a single layer of cuboidal cells encased by a basement membrane, which is referred to as the lens vesicle. This basement membrane is the future lens capsule. The cells in the posterior cellular layer stop dividing, elongate, and lose their organelles. At approximately 40 days, the lumen of the lens vesicle is filled with these primary lens fibers. This structure is now the optically clear embryonic nucleus. The anterior layer of cells remains a monolayer of cuboidal cells referred to as the lens epithelium. The secondary lens fibers originate from the anterior epithelium in the equatorial region. These cells migrate anteriorly and posteriorly beneath the capsule. The fibers meet and interdigitate to form the Y sutures. The anterior suture has the appearance of an upright Y, while the posterior suture has the appearance of an inverted Y. The secondary lens fibers are formed between 2 and 8 months of gestation and form the fetal nucleus. The adult nucleus and cortex are formed by the continued proliferation of the equatorial lens epithelium.
The lens fibers continue to be added throughout life. At birth, the lens weighs approximately 90 mg and its mass increases by approximately 2 mg per year as new fibers are formed. The average diameter of the lens at birth is 7.0 mm. The most rapid increase in the size of the lens occurs during the first 2 years of life. The average diameter of the lens at age 2 is approximately 9.5 mm (
4). Because the diameter of the lens is similar to the adult size early in life and the fact that lens fibers continue to be added throughout life, the density of the lens increases with age. It should also be noted that the lens capsule continues to thicken throughout life.
LENS OPACITIES
The most common lens abnormality in children is opacity (cataract). The visual significance of the cataract depends on several factors including age of onset, location, etiology, and morphology.
Etiology of Childhood Cataracts
The etiology of childhood cataracts is vast (
Table 11.1). The historical teaching is that approximately one-third of childhood cataracts are inherited, one-third are associated with other diseases or syndromes, and one-third are idiopathic.
Hereditary Cataract
Hereditary cataracts account for a significant proportion of childhood cataracts. Inheritance patterns that have been reported include autosomal dominant, autosomal recessive, and X-linked recessive. The most common form of inheritance is autosomal dominant. There is often high penetrance with phenotypic variability. These children are usually otherwise normal. Less commonly, autosomal recessive cataracts occur. They are usually bilateral but can also show variability between affected family members.
Metabolic Cataract
Errors in metabolism may cause cataracts in neonates, infants and juveniles. Although lens opacities may be permanent, occasionally the early recognition and treatment of underlying disease may reverse the lens damage. It is also critical to recognize and treat the underlying metabolic etiology early for the child’s overall health and well-being.
Galactosemia
Galactosemia is an autosomal recessive inherited condition where the child cannot metabolize galactose, a major component of milk. There is a defect in galactokinase, uridine diphosphate galactose-epimerase, or galactose 1-phosphate uridyltransferase. Because of this defect, galactose is converted to galactitol in the crystalline lens, which makes the lens hyperosmolar, resulting in an influx of water into the lens. The hydration of the lens disrupts the normal packing of the lens fibers, and transparency is lost. The typical lens change described in galactosemia is the “oil droplet,” which is not a true cataract but represents refractive change within the lens. The appearance is that of a drop of oil floating in water. If the baby remains untreated, a diffuse white lens will develop. If the baby is diagnosed early, within the first weeks of life, the lens changes are reversible. Unfortunately, this is not always the case, and surgery may be required to treat the cataracts. It is important to note whether the baby has other systemic issues including: vomiting, diarrhea, failure
to thrive, and hepatomegaly. If the baby remains untreated, mental retardation and death may occur. Screening for galactosemia is now included in the newborn screening mandatory in many states. Finally, galactosemia is a life long condition, so these children must be followed throughout life for development of cataracts even though they are following dietary restrictions.
Fabry’s Disease
Fabry’s disease is an uncommon, X-linked recessive metabolic disorder caused by a deficiency of the lysosomal enzyme α-galactosidase A. Abnormal storage of glycosphingolipids causes a chronic progressive painful small-fiber neuropathy, renal dysfunction, heart disease, and stroke. In males, symptoms often start in adolescence with pain in the extremities provoked by exertion or temperature changes. The main lifelimiting complications are renal disease and heart disease. Ocular involvement includes the conjunctiva, cornea, lens, and retina. There are two types of lens opacities associated with Fabry’s disease. The first is a spoke-like posterior line of spots that radiate and are best seen in retroillumination. The second is an anterior subcapsular, wedge-shaped opacity usually in the inferior lens (
5).
Mannosidosis
Mannosidosis is an autosomal recessive disorder resulting from a deficiency in α-mannosidase, which causes defective degradation of lipoproteins. Patients have coarse facies, skeletal abnormalities, deafness, and learning difficulties. Cataracts are common and present early in life. They appear as posterior cortical spokes composed of discrete clear vacuoles (
6).
Wilson’s Disease
Wilson’s disease is an autosomal recessive disorder caused by a deficiency of an ATPase that transports copper into the Golgi apparatus of the hepatocytes. This leads to the toxic accumulation of copper in the hepatocytes and elsewhere. The plasma concentration of non-ceruloplasmin-bound copper is elevated, and copper deposits occur in the liver, brain, and eye. As the lens accumulates copper, a unique sunflower cataract forms, which is a yellowish star like anterior subcapsular discoloration.
Hyperglycemia and Hypoglycemia
Hyperglycemia and hypoglycemia are infrequent causes of cataracts in children. Hypoglycemia in the perinatal period is common in low-birth-weight infants. Lens opacities may result and are often bilateral and lamellar in type. These opacities are often reversible but may progress into total cataracts. Cataracts in diabetic children do occur but often not until the teenage years. They are often diffuse, cortical, or subcapsular in morphology.
Disorders of Cholesterol Synthesis
Because the lens membrane contains the highest cholesterol content of any known membrane, inherited defects in enzymes of cholesterol metabolism are associated with cataracts. Smith-Lemli-Opitz syndrome, mevalonic aciduria, Conradi-Hünermann syndrome, and cerebrotendinous xanthomatosis all have mutations in enzymes of cholesterol metabolism, and cataracts have been reported in all of them. The pathogenesis of the cataracts in this group of disorders remains unknown (
7).
Hypocalcemia
Children with hypocalcemia may present with irritability, failure to thrive, and seizures. Cataracts have been reported in children with hypocalcemia and are thought to occur because of altered permeability of the lens capsule. They start as fine white punctuate opacities scattered throughout the lens cortex, which are not visually significant but progression may occur to form lamellar cataracts that become visually significant.
Cataract Associated with Systemic Syndromes
There are many multisystem disorders that have cataracts as part of the syndrome. The incidence of cataracts in this group varies widely. The following text outlines a subset of entities in which cataracts are found in a high percentage of involved patients.
Lowe’s Syndrome
Lowe’s Syndrome (oculocerebrorenal) is an X-linked recessive condition in which almost 100% of patients have cataracts. Children with Lowe’s syndrome have mental retardation, hypotonia, renal aminoaciduria, and typical facies. The lens in Lowe’s syndrome is usually a small, flat, disklike opacified lens. Glaucoma and corneal opacities are common. Of interest, female carriers often have lens opacities that are diffuse punctate opacities or spoke-like opacities in the posterior cortex.
Alport’s Syndrome
Alport’s syndrome is an X-linked or autosomal recessive disorder consisting of interstitial nephritis, hearing defects, and ocular abnormalities. The lens abnormality is that of anterior lenticonus, which is secondary to an abnormally thin central anterior capsule. Cataract is usually a late finding in anterior lenticonus, but surgery may be required earlier because the thinning of the capsule causes significant aberration of the optics of the natural lens. Rarely, a total cataract may occur if there is spontaneous rupture of the anterior lens capsule.
Chondrodysplasia Punctata
This is a group of disorders, which is broken down into chondrodysplasia punctata and rhizomelic chondrodysplasia
punctata. Each of these is then broken down into type one and type two. Chondrodysplasia punctata 1 is X-linked recessive affecting chromosome Xp22.33 and is caused by mutation in the arylsulfatase E gene. These children have short stature, hearing loss, skeletal abnormalities, ichthyosis, and developmental delays. Chondrodysplasia punctata 2 (Conradi’s syndrome) is X-linked dominant affecting chromosome Xp11.23 and is caused by defect in cholesterol synthesis. These children have failure to thrive, hearing loss, tracheal stenosis, many skeletal abnormalities, ichthyosis, and mental retardation. The rhizomelic forms are autosomal recessive with defects in the peroxisomal biogenesis pathway. Cataracts are reported in up to 75% of the rhizomelic chondrodysplasia infants, and early death is expected. All of the forms of chondrodysplasia punctata have related cataracts (
8).
Myotonic Dystrophy
Myotonic dystrophy is an autosomal dominant muscular dystrophy, which presents with progressive muscle weakness and muscle wasting. Other systemic features include gonadal atrophy, frontal balding, mental deterioration, and cardiac abnormalities. Ocular features include ptosis, progressive external ophthalmoplegia, retinal pigmentary changes, and cataracts. Typical lens changes occur in almost all patients and present as multicolored crystalline iridescent flecks in the cortex and are often referred to as a “Christmas tree” cataract. The lens opacities may also present as small white spherical opacities in the cortex.
Neurofibromatosis Type 2
Neurofibromatosis type 2 is an autosomal dominant trait with high penetrance, which is characterized by vestibular schwannomas and other central nervous system tumors. Cataracts are often present with the most common type being the posterior subcapsular and cortical cataract.
Zellweger Syndrome
Zellweger syndrome is an autosomal recessive disease which consists of abnormal development of the head, face, ears, and feet. These patients also have mental retardation, hypotonia, seizures, and early death. Lamellar cataracts are present in most cases. Other ocular findings include corneal clouding, pigmentary retinopathy, optic atrophy, and glaucoma. Asymptomatic carriers have been found to have curvilinear opacities. This disorder is caused by an error in peroxisome biogenesis involving a defect in the PEX group of genes (
9).
Cockayne Syndrome
Cockayne syndrome is an autosomal recessive disorder with a defect of transcription-coupled DNA repair of active genes. The most striking feature of this disorder is premature aging and cachectic dwar. sm. Almost all organ systems are affected. There is progressive neurologic deterioration, mental retardation, skeletal abnormalities, skin photosensitivity, and sensorineural hearing loss. Ocular anomalies include cataracts, retinal dystrophy, corneal opacity, decreased lacrimation, optic atrophy (
10).
Bloch-Sulzberger Syndrome
Bloch-Sulzberger syndrome (incontinentia pigmenti) is an X-linked dominant disease affecting the skin, bones, teeth, central nervous system, and eyes. The disorder is usually lethal in males. The skin changes go through 4 distinct stages starting with linear bullae and ending in atrophy and scarring. Ocular anomalies are numerous and include cataract, optic atrophy, microphthalmos, retinal changes leading to retinal detachment, and epithelial keratitis. The lens abnormality is reminiscent of that seen with persistent fetal vasculature.
Rothmund-Thomson Syndrome
Rothmund-Thomson syndrome is an autosomal recessive condition consisting of atrophic skin with patches of depigmentation and hyperpigmentation. Patients have telangiectasias, sparse hair, short stature, defective dentition, and hypogonadism. Lamellar cataracts occur in the majority of patients and often have an onset between 3 and 6 years of age.
Hallermann-Streiff Syndrome
Hallermann-Streiff syndrome is an isolated disease, which involves dyscephaly with a beak-shaped nose and micrognathia, short stature, dental abnormalities, respiratory issues, multiple skeletal anomalies, and congenital cataracts. The eyes in Hallermann-Streiff are usually microphthalmic. The lens in Hallermann-Streiff syndrome may be small and has been reported to spontaneously resorb to form a disk-like or membranous type cataract.
Cataract Associated with Chromosomal Anomaly
Cataracts are present in a large number of chromosomal syndromes with the most common being trisomy 21. Cataracts in Down syndrome usually develop later in childhood but they may be present during infancy. Cataracts are also part of the clinical presentation in trisomy 13, trisomy 15, and trisomy 18. Cataracts have also been reported in chromosome deletions with the most common being Turner syndrome (XO) and Cri-du-chat (5p-). Finally, cataracts are part of several translocation defects such as 3:4 translocation, 2:14 translocation, and 2:16 translocation. As genetic mapping continues, the number of known chromosome anomalies associated with cataracts will increase.
Secondary Cataracts
Secondary cataracts can be thought of as lens opacities caused by external forces or secondary to other ocular process.
Maternal Infection
Many intrauterine infections can cause cataracts. The cataracts are usually central and can be bilateral or unilateral. The most common cause of cataract from maternal infection is rubella. Systemic manifestations of congenital rubella infection include cardiac defects, mental retardation, and deafness. The cataract is usually a pearly white nuclear opacity. Sometimes the entire lens is involved and becomes white, the cortex may even liquefy. The lens does contain live virus, and removal is often associated with difficult-to-control postoperative inflammation. Rubella cataracts are still an important cause of cataracts worldwide but are rare in the United States. Cataracts can also occur in toxoplasmosis, varicella, cytomegalovirus, and toxocariasis intrauterine infection.
Drug Toxicity
The lens can be sensitive to both topical and systemic medications. The most common drugs known to cause cataract formation are the corticosteroids. This effect appears to be dose and duration related. The type of cataract is usually posterior subcapsular, but can progress to involve the entire lens.
Iatrogenic Cataracts
Development of cataract related to radiation is seen in children who have had total body radiation in preparation for bone marrow transplant. These cataracts are usually posterior subcapsular and are dose and duration dependent. The incidence of cataract increases with increasing dose of radiation. Cataracts have been reported after laser treatment for threshold of retinopathy of prematurity. Finally, any intraocular surgery increases the risk of cataract formation in children.
Cataract Secondary to Ocular Process
The most common type of secondary cataract related to an intraocular process is uveitis. The uveitis may be anterior, intermediate, or posterior in nature. The cataract may be related to the inflammation or a result of the steroids used to treat the inflammation. Less common associations are with intraocular tumor, intraocular foreign body, or chronic retinal detachment.
Traumatic Cataracts
Traumatic cataracts are a frequent and important cause of cataracts in children. The cataract may be secondary to penetrating, perforating, or blunt trauma. The cataract may form immediately after injury or be delayed in its formation as can be seen in blunt force trauma. Cataracts secondary to trauma may be partial or complete. It is important to be aware that both accidental and inflicted trauma may be the cause. Surgical intervention is only required for visually significant cataracts. The visual prognosis of traumatic cataract is dependent on accompanying ocular injuries.
Cataracts Associated with Ocular Anomalies
Cataracts have been associated with many ocular anomalies including microphthalmia, aniridia, retinitis pigmentosa, and coloboma. The cataract associated with persistent fetal vasculature deserves special mention. This ocular condition is caused by a failure of the primitive hyaloid vascular system to regress and is most commonly unilateral. The ocular abnormalities most commonly consist of a vascularized retrolental plaque in a microphthalmic eye with prominent iris blood vessels, shallow anterior chamber, and elongated ciliary processes. The lens is often clear initially but opacifies with time. The lens may also move forward with time and glaucoma may develop. The retina may be involved in two ways. There may be contraction of the retrolental plaque causing traction on the vitreous base and peripheral retina. If a remnant of the hyaloid vessel is present at the optic nerve, it may become fibrous causing peripapillary retinal folds or detachment. Treatment involves removing the lens and the fibrovascular membrane. It is often difficult to remove the membrane as it is thick and difficult to cut with the vitrector. The hyaloid artery may still have blood in it and may require cauterization at the time of surgery to avoid vitreous hemorrhage. Finally, the peripheral lens material must be removed carefully to avoid damaging the ciliary processes. Visual outcomes may be affected by amblyopia, glaucoma, retinal folds, and retinal detachment.
Cataracts of Unknown Etiology
Many surgeons feel that a majority of nontraumatic unilateral cataracts are idiopathic in nature, but bilateral cataracts may also be idiopathic. A complete ocular history and examination must be performed with detail directed to history and findings consistent with trauma or inflammation. An ocular examination of the parents may reveal visually insignificant cataracts that the parent did not know were present. Further workup may be indicated in some children but should be done with the assistance of a developmental pediatrician or geneticist to focus the testing for improving the likelihood of a positive result (
Tables 11.2 and
11.3).
Morphology of Pediatric Cataracts
The morphology of childhood cataracts can be an important tool in determining the age of onset of the cataract, the possible etiology of the cataract, and visual prognosis. The morphology of the cataract is determined by the anatomy of the lens, lens embryology, and the timing and nature of the
insult. Some morphological types have better prognosis, so trying to identify and classify the cataract may be a helpful management tool. The following classification of morphology is broken down into four types mainly based on location of the opacity: total, central, anterior, and posterior.
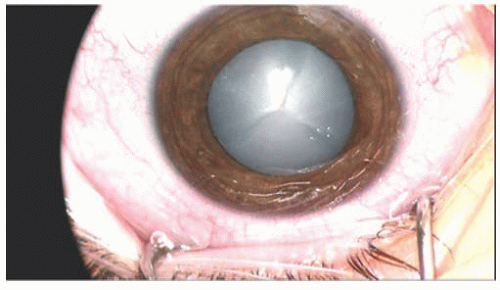
Cataract Involving the Whole Lens
Total Cataract A total cataract is complete opacification of the whole lens (
Fig. 11.1). They can be caused by any number of conditions and is not diagnostic of any one disorder. Many types of cataracts may progress to total
cataracts if left untreated. Because no view of structures posterior to the lens is possible, b-scan ultrasonography is recommended to evaluate for retinal detachment, intraocular tumor, or intraocular foreign body. These cataracts require surgical intervention as they are all visually significant.
A special type of total cataract is the Morgagnian cataract. In this cataract the lens fibers liquefy, but the nucleus remains intact. The nucleus will then move within the lens capsule, depending on gravity. These types of cataracts are uncommon in the United States.
Membranous Cataract The membranous cataract is a thin fibrotic lens caused by resorption of the lens material. The anterior and posterior capsules fuse to form a dense white membrane. This type of cataract has been associated with trauma, posterior or anterior capsule defects, congenital rubella, Hallermann-Streiff syndrome, persistent fetal vasculature, Lowe’s syndrome, and aniridia (
11).