Pathology of the Uvea
P. Kumar Rao
The uvea is the pigmented vascular middle layer of the eye, lying between the sclera and neuroepithelium. It consists of three parts: the iris, which is the anterior part of the uvea; the ciliary body, forming the middle; and the choroid, which is the posterior section. Common pathologic changes involving the uvea include inflammatory and neoplastic diseases. Inflammatory changes are clinically recognized as various forms of uveitis. Among the neoplasms, both primary and metastatic tumors are found in all parts of the uvea.
EMBRYOLOGY
Neural crest cells are responsible for the development of many structures of the eye and orbit.1,2 Anterior segment development results from three waves of neural crest cell migration between the surface ectoderm and lens vesicle.3 The first wave of migrating neural crest cells gives rise to trabecular meshwork and endothelium. The second wave becomes keratocytes, and the third wave differentiates into iris stroma.3 The muscular elements of the iris and its pigment epithelium derive from the neural epithelium. The neural epithelium is also responsible for the development of the neural (sensory) retina and the retinal pigment epithelium. Other derivatives of neural crest are pigmented and nonpigmented portions of iris and choroidal stroma, scleral fibroblasts, and many adnexal structures.4,5,6 Various anterior segment disorders can be explained by abnormal neural crest development (Table 1).
Table 11-1. Classification of Anterior Segment Disorders Based on Neural Crest Origin | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Reciprocal cellular interactions are particularly important in the differentiation of the ciliary body and iris.7 Lens epithelium must interact with the potential ciliary epithelium and iris to differentiate properly.7 Without the developing lens, the neural plate epithelium that would have become ciliary epithelium differentiates into retina.8 Ciliary epithelium in turn induces underlying neural crest tissue to differentiate into ciliary muscle and stroma.7
The choriocapillaris begins to differentiate at the fourth or fifth week of gestation at approximately the same time that the retinal pigment epithelium begins its differentiation. In this way the developing human eye is completely enveloped in a capillary layer by the sixth gestational week. The external, large Haller vessels develop during the fourth month of gestation. They consist of branches of short posterior ciliary arteries and veins draining the choriocapillaris. Sattler vessels develop during the fifth month between the choriocapillaris and Haller layer of vessels. By week 23, all choroidal vascular layers are present, and arteries, as distinct from arterioles, are present.9,10
CONGENITAL AND DEVELOPMENTAL ABNORMALITIES
Coloboma
Coloboma, a localized defect within tissue, may involve any part of the uveal tract. Typical colobomas occur inferonasally, in the region of the embryonic fissure. They may be complete or incomplete (iris stromal hypoplasia) or, when in the region of the choroid, may appear cystic. Atypical colobomas occur in regions other than the inferonasal area. After initial formation of the optic vesicles from neuroectoderm, invagination of the optic vesicle occurs and forms the optic cup. The embryonic fissure forms secondary to progressive invagination along the inferior edge of the optic cup and stalk. Prior to closure of this fissure, it receives the ingrowth of the hyaloid artery and permits retinal axons to form the optic nerve. Closure of the embryonic fissure begins in the middle and progresses proximally and distally. Improper fusion of the inner optic cup layers leads to failure of the outer layers to become confluent, resulting in a coloboma. The embryonic fissure is located in the inferonasal region of the eye, and failure of fusion leads to typical colobomas in that area. The embryonic fissure closes by 33 to 40 days of embryonic life.11,12
Histologically, a simple chorioretinal coloboma consists of bare sclera that is not covered by choroid or normal retina. The sclera may be covered by a membrane of undifferentiated retina that may contain blood vessels.11
Ocular coloboma usually is sporadic or inherited as an autosomal dominant disorder that is not associated with extraocular abnormalities. Recessive inheritance also has been reported.11 Coloboma may be a component of many syndromes. Only a few coloboma-associated syndromes of particular interest to ophthalmologists are discussed here.
CHARGE syndrome consists of coloboma, heart defects, atresia of choanae, retarded growth and development, genital hypoplasia, and ear abnormalities or hearing loss.13 Chestler and France13 found that in 54 cases of ocular coloboma, 6 patients (11%) met the diagnostic criteria for CHARGE syndrome. Patients had vision of 20/200 or less in at least one eye, microphthalmos, iris and chorioretinal defects, and optic nerve colobomas. Sixty to seventy percent of CHARGE syndrome cases may be caused by a mutation in the CHD7 gene, which encodes for a chromodomain helicase DNA binding protein. These mutations may be inherited in an autosomal dominant fashion.14
Patients with Aicardi syndrome also suffer from colobomas. Font et al15 have documented the ocular abnormalities in Aicardi syndrome. Findings included bilateral microphthalmos, bilateral optic nerve hypoplasia, bilateral colobomas of the juxtapapillary choroid and optic disc, bilateral total retinal detachment with dysplastic rosettes and chorioretinal lacunae characterized by focal thinning, and atrophy of the retinal pigment epithelium and choroid.15
IRIS
Normal Anatomy
The iris is made up of three layers: an anterior layer composed of fibroblasts, melanocytes and collagen; a middle layer of stroma; and a posterior layer composed of the dilator muscle and pigment epithelium. The anterior layer terminates at the iris root with the exception of some spoke-like extensions that continue into the Schwalbe line, and its density determines iris color. The stroma makes up the bulk of the iris and consists of pigmented and nonpigmented cells in a loose extracellular matrix of collagen and mucopolysaccharides. There is a rich blood supply to this layer, in addition to nerves. In the pupillary zone, the iris sphincter muscle is present. Unlike the anterior layer, the stroma appears to have a similar structure regardless of its iris color. In the posterior layers, the dilator muscle extends from the pupillary zone to the periphery. The pigment epithelium is composed of two layers of apposed epithelia which are arranged apex to apex. The anterior cuboidal border is continuous with the pigmented epithelium of the ciliary body. The posterior epithelial cells are columnar.16
Aniridia
Aniridia describes a clinical appearance of complete iris hypoplasia. In actuality, a small stump of iris is present on gonioscopic examination as well as histologically. Most cases are familial and are usually inherited in an autosomal dominant manner.17 The condition may be accompanied by glaucoma, attributable to a progressive synechiae of the trabecular meshwork and iris remnant.18 An association of Wilms tumor, aniridia, genitourinary abnormalities, and mental retardation is known as the WAGR syndrome.19,20 A deletion in the 11p14-p12 region may account for the this syndrome.21,22
Axenfeld-Reiger Anomaly
In the Axenfeld-Reiger anomaly there is thickening and anterior displacement of the Schwalbe line. Iris stromal processes may be adherent to this abnormally thickened Schwalbe line.
Iridocorneal Endothelial (ICE) Syndrome
The ICE syndrome refers to a set of acquired abnormalities affecting the cornea, angle, and iris. This syndrome may be subdivided into iris nevus syndrome, Chandler syndrome, and essential iris atrophy. Abnormal proliferation of the corneal endothelium is a feature of this syndrome, as well as production of a basement membrane.23,24 Adhesion of the iris stroma to the endothelium result in peripheral anterior synechiae with progression to angle-closure glaucoma. Each condition demonstrates endothelial attenuation and extension with production of a basement membrane. If the basement membrane grows over the trabecular meshwork, endothelialization and descemetization of the chamber angle may be seen. As the endothelium extends over the trabecular meshwork, changes in the iris result. Specular microscopy has demonstrated pleomorphism of the endothelium early in the disease.25,26 Histologically, the endothelial cells may show pleomorphism, anisocytosis, and occasional paired oval nuclei.23 In vivo confocal microscopy of affected patients may reveal similar pleomorphic changes and paired oval nuclei as well as hyperreflective nuclei.27
Iris Cysts
Iris cysts can be classified based on their location.28 In general, iris cysts are of the pigment epithelium or of the iris stroma. Most iris epithelial cysts remain stable and do not progress. If the cyst is located in an area of iris pigment epithelium, the cyst will fail to transilluminate. In contrast, if the iris cyst is more peripheral and located near the ciliary body nonpigmented epithelium, the cyst may allow transillumination. As its name implies, a cyst of the pigment epithelium is lined by the iris pigment epithelial cells. On the other hand, cysts of the iris stroma are lined by stratified squamous epithelium with goblet cells and may resemble conjunctival epithelium (Fig. 11-1). These findings and the fact that stromal cysts are usually found in infants support the idea of a congenital rest of ectopic surface epithelium as the source of these lesions.
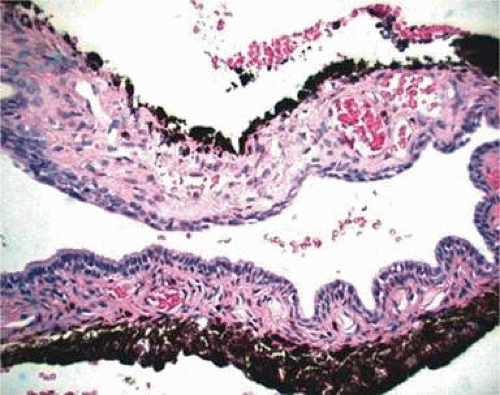 Figure 11-1. Iris cyst. Iris stromal cyst lined by stratified epithelium that resembles conjunctival epithelium. (Hematoxylin and eosin ×25.) |
Acquired cysts of the iris may be due to epithelial down-growth in postsurgical or traumatic cases, intraocular tumors such as medulloepithelioma, or even the use of topical prostaglandin analogues used in the therapy of glaucoma. Additionally, some parasitic infections such as cysticercosis may present with cystic changes.29
Iris Nodules
Iris nodules may appear in many conditions and have different appearances based on their etiology. Etiologies include congenital diseases as well as inflammatory, neoplastic, and infectious causes. Congenital accumulations of nevus cells may be variable in their pigmentation. These nevi may be seen alone or in association with neurofibromatosis.30 An iris freckle is seen as a darkly pigmented flat area on the anterior iris surface and is composed of melanocytes containing an increased amount of pigmentation without an increase in the number of cells. Brushfield spots seen in Down syndrome appear as elevated white-to-yellow lesions in the periphery of the iris and histopathologically appear as areas of relatively normal iris stroma surrounded by a ring of mild iris hypoplasia.31 In the iris nevus (Cogan-Reese) syndrome, diffuse nevi of the iris associated with unilateral glaucoma and peripheral anterior synechia may be seen in this variant of the ICE syndrome.
Inflammatory conditions may also lead to iris nodules. Patients suffering from fungal endophthalmitis may demonstrate an irregular yellow-white mass on the iris. Histologically, these appear as necrotizing granulomas containing mycotic agents (Fig. 11-2). In juvenile xanthogranuloma, a yellowish-gray iris lesion may be associated with spontaneous hyphema, and histopathologically the nodules demonstrate diffuse histiocytic infiltrate (Fig. 11-3). Multinucleated giant cells displaying peripheral foamy cytoplasm are also noted; these cells are known as Touton giant cells.32 The giant cells and the histiocytes contain lipid that can be demonstrated by oil red O stain.
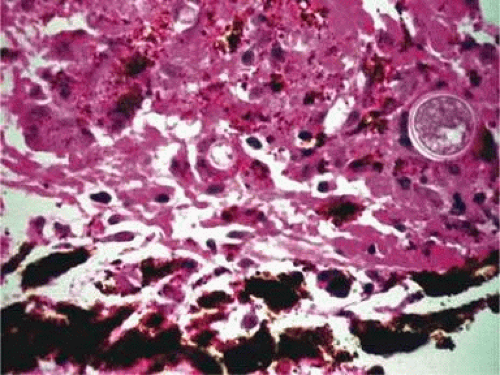 Figure 11-2. Coccidioidomycosis. Iris stroma shows necrotizing granuloma containing mycotic organisms. The organisms show features of Coccidioides immitis. (Hematoxylin and eosin ×60.) |
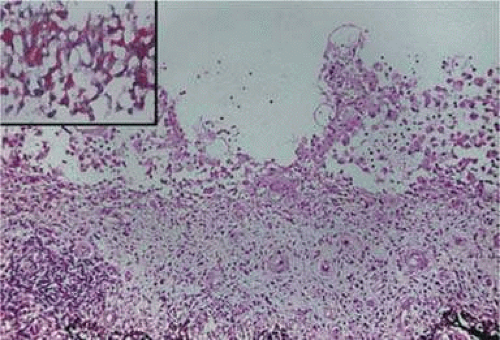 Figure 11-3. Juvenile xanthogranuloma. The iris is infiltrated by histiocytes, which form nodular aggregates on the anterior surface of the iris. (Hematoxylin and eosin ×25.) Inset (×200) shows oil red O-positive histiocytes. |
Patients suffering from classic granulomatous anterior uveitis may demonstrate Koeppe nodules that occur at the pupillary border, or Busacca nodules at the anterior iris surface. Such nodules are seen in patients with sarcoidosis, tuberculosis, Vogt-Koyanagi-Harada disease, and others. Histologically, they consist of aggregates of epithelioid cells mixed with other mononuclear cells. However, in tuberculosis the granulomas show necrosis, and acid-fast stains may reveal the mycobacteria.
Neoplastic causes of iris nodules include melanoma, leiomyoma, leukemia, metastatic carcinoma, and retinoblastoma. Melanoma may occur as a nodular (Fig. 11-4A) or flat growth or as tapioca-like nodules. All these lesions show spindle-shape tumor cells (Fig. 11-4B), or such cells may be mixed with epithelioid melanoma cells. Both spindle A and spindle B cells may be present.33 These tumors may display occasional mitotic figures, foci of necrosis, and melanophages. Iris melanomas usually are small, mainly low-grade spindle tumors and carry a relatively good prognosis when compared to ciliary body and choroidal melanomas.34,35,36,37,38,39,40,41,42
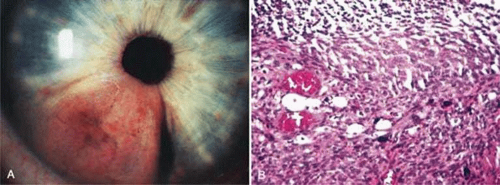 Figure 11-4. Iris melanoma. A pigmented mass involves the iris (A), which histologically reveals spindle cells (B). (Hematoxylin and eosin ×60.) |
Histologically, leiomyoma may appear similar to amelanotic spindle cell melanoma; immunohistochemical analysis with anti–muscle-specific actin and electron microscopy may be required for clear differentiation.43,44
In leukemia, rarely nodular or milky lesions may be present. Histologically, the nodules reveal infiltration of abnormal lymphoid (Fig. 11-5) or myeloid cells and iris architecture is lost. As the lesion becomes thickened, a pseudohypopyon is common.45 In cases of retinoblastoma, white foci on the anterior iris surface may appear and a pseudohypopyon may also be present.46 Histologically, these foci show abnormal, hyperchromatic, round cells with scant cytoplasm as well as frequent abnormal mitotic figures. Adenomas of the pigmented iris epithelium and adenomas of nonpigmented or pigmented ciliary epithelium may present with nodular lesions pushing the iris stroma. These benign tumors rarely enlarge, seldom undergo malignant transformation, and consist of proliferations of the iris or ciliary body epithelium. Other rare tumors include neonatal hemangiomatosis, and this entity shows multiple vascular spaces that contain erythrocytes and are lined by endothelium (Fig. 11-6).
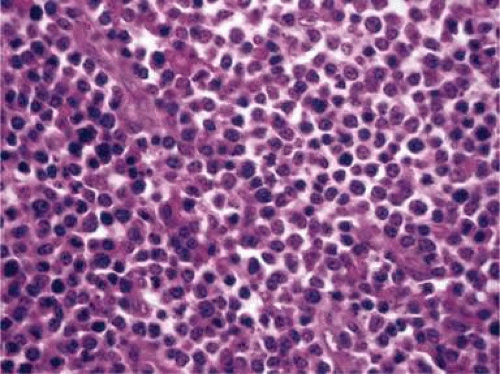 Figure 11-5. Lymphoma of the iris. Iris stroma is infiltrated by monomorphic-appearing lymphoid cells. The tumor cells were positive for T-cell marker CD3. (Hematoxylin and eosin ×60.) |
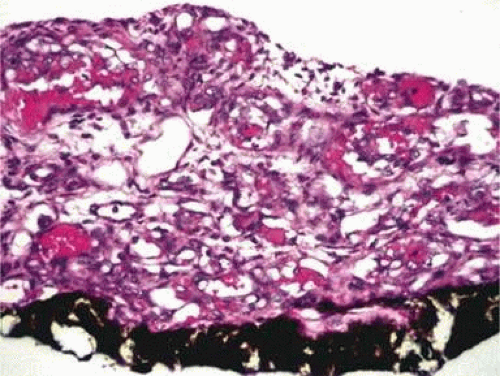 Figure 11-6. Neonatal hemangiomatosis. Multiple vascular spaces lined by endothelial cells; some of the spaces contain erythrocytes. (Hematoxylin and eosin ×25.) |
Diabetes
Iris changes resulting from diabetes can be seen histopathologically in proliferative diabetic retinopathy. Neovascularization may be seen on the anterior surface of the iris and may contract and pull the pigmented iris epithelium around the pupillary edge, creating ectropion uvea. Additionally, the neovascular membrane may grow over the trabecular meshwork, leading to neovascular glaucoma (Fig. 11-7A). Lacy vacuolization of the iris pigment epithelium may be seen histologically if enucleation is performed during a hyperglycemic state. These intraepithelial vacuoles contain glycogen and are periodic acid–Schiff (PAS) positive. In addition, the basement membrane of the ciliary pigment epithelium may show diffuse thickening (Fig. 11-7B).47
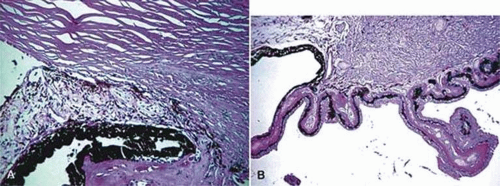 Figure 11-7. A. Neovascular glaucoma. The anterior chamber angle is blocked by peripheral anterior synechia. Neovascularization of the iris is present. (Hematoxylin and eosin ×25.) B. Diabetes. The ciliary epithelium shows diffuse thickening of the basement membrane. (Periodic acid–Schiff ×35.) |
CILIARY BODY
Normal Anatomy
The ciliary body consists of an anterior portion called the pars plicata and a posterior portion called the pars plana. The stroma of these areas are filled with melanocytes, rich vascular networks, fibrous connective tissue, and bundles of smooth muscle. The smooth muscle constitutes most of the ciliary body and can be divided into three portions: an outer longitudinal portion, a middle oblique portion, and the inner circular component. The ciliary muscles connect to the scleral spur just posterior to the trabecular meshwork. The ciliary body is covered by a bilayered epithelium; the outer layer of this epithelium is pigmented, but the inner layer is not. The pars plicata is denoted by multiple finger-like extensions of ciliary body stroma covered by the ciliary body epithelium. The pars plana does not have these extensions and ends at the ora serrata. In children, the processes of the ciliary body are thin and finger-like. With age, these ciliary processes become thickened and show eosinophilic acellular material.
Pathologic changes involving the ciliary body can be divided into those related to congenital abnormalities and others related to acquired changes. The latter include pathologic alterations resulting from trauma, inflammatory and infectious changes, and neoplastic and degenerative changes.
Congenital Abnormalities
Iris and Ciliary Body Cysts
In a review of patients with primary iris cysts, Shields et al48 studied 45 patients with cysts located near the junction of the iris and ciliary body. These cysts tended to be unilateral, affected women, and were usually located in the temporal quadrants. Most cysts remained stable and were not associated with anterior segment complications such as glaucoma, cataract, or corneal edema. Histologically, these cysts were located where the ciliary body’s nonpigmented epithelium joined the iris pigment epithelium, and the presence of some nonpigmented epithelium in the cyst wall explained why these cysts would transilluminate. Some of the cysts are lined by pigment epithelium (Fig. 11-8).
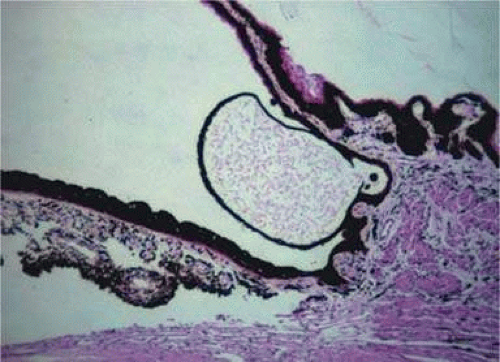 Figure 11-8. Ciliary epithelial cyst. The cyst arising from the ciliary epithelium is lined by pigment epithelial cells. (Hematoxylin and eosin ×25.) |
Stratified squamous epithelium with goblet cells was found in primary stromal cysts and may indicate a congenital rest of ectopic surface epithelium as the source of these lesions. In a study of 232 presumed normal eyes, 54% had ciliary body cysts when examined with ultrasound techniques. There was no gender predominance in this study.49 Congenital cysts of the vitreous have also been reported.50,51 Their origin is unknown. Cysts in the anterior vitreous may lie behind the lens or ciliary body and may be connected by thread-like extensions. It is possible that these anterior cysts may be derived from ciliary body epithelium.
Persistent Hyperplastic Primary Vitreous, Trisomy 13–15
Persistent hyperplastic primary vitreous (PHPV) is usually a unilateral condition that leads to fibrovascular connections in the retrolental area. These connections may reach the posterior retina and cause a severe tractional detachment. Adipose tissue, cartilage, and smooth muscle may be seen in the retrolental mass. Ciliary processes are stretched or elongated as well.52,53,54,55,56,57 A group of disorders characterized by trisomy 13–15 may also demonstrate the elongated ciliary processes and retrolental findings seen in PHPV.58,59,60 This condition (also termed Patau syndrome) demonstrates retinal dysplasia and rosette-like configurations of the retina.
Acquired Abnormalities
Cysts of the Pars Plana
Pars plana cysts appear to be acquired rather than congenital and may represent a separation of the two epithelial layers analogous to retinal detachments seen posteriorly. They are presumed to be a degenerative change. Histologically, they appear as intraepithelial cysts within the nonpigmented ciliary epithelium of the pars plana.61,62,63 They appear empty in routinely stained sections but have been shown to contain hyaluronidase-sensitive material, presumably hyaluronic acid.62
Cysts of the pars plana have been demonstrated in patients with multiple myeloma. In contrast to typical pars plana cysts, those associated with myeloma become opaque when fixed in formaldehyde and are eosinophilic on hematoxylin and eosin stain. Additionally, they are PAS positive.61,64,65 These cysts have been demonstrated to contain a gamma globulin and may be seen in patients with nonmyelomatous hypergammaglobulinemia.66
Trauma
Following blunt injury to the eye, a cleft between the circumferential and longitudinal ciliary body muscles may develop (Fig. 11-9). This is described clinically as a recessed angle. Histologically, one can determine angle recession by tracing an imaginary line through the scleral spur and parallel to a line passing between the pupil and optic nerve. If the ciliary body is posterior to this line, the angle can be termed recessed. Complete dislocation of the ciliary body from the scleral spur is known as cyclodialysis.67
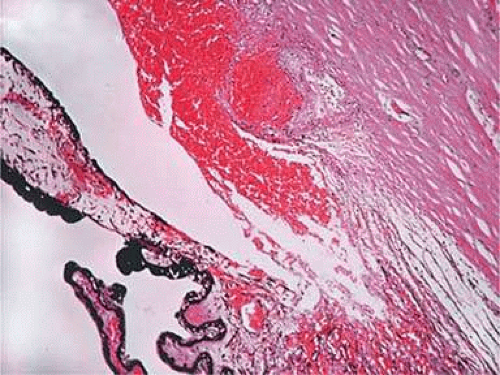 Figure 11-9. Ciliary body with acute angle recession. There is a cleft between the ciliary muscle and hemorrhage involving angle structures. (Hematoxylin and eosin ×25.) |
The uveitis/glaucoma/hyphema (UGH) syndrome represents episodes of recurrent hemorrhage associated with uveitis and elevated intraocular pressure following implantation of an intraocular lens. The condition is thought to result from repeated trauma to the iris and ciliary body structures from the implanted intraocular lens. Erosion of the posterior chamber intraocular lens haptics into the substance of the ciliary body can occur.68,69
Inflammatory and Infectious Changes
Intermediate Uveitis (Chronic Cyclitis, Pars Planitis)
The pars plana and peripheral choroid can be involved with nongranulomatous chronic inflammatory infiltrate. Clinically, such inflammation is seen as vitritis and whitish exudative changes at the pars plana. When it is idiopathic, such inflammatory infiltrate may be termed pars planitis. A moderate to dense vitritis may be present. The vitreous inflammation may clump together and form “snowballs.” Typically, dense white material is present at the pars plana and posterior ciliary body; this is called a “snowbank.” Histologically, enucleated eyes with pars planitis reveal a lymphocytic infiltration in the pars plana and peripheral choroids (Fig. 11-10). The snowbank is made of fibroglial proliferation containing elements of ciliary epithelium, and vascular channels surrounded by mononuclear cells.70,71,72 Features similar to pars planitis are also observed in patients with multiple sclerosis. Intermediate uveitis without a clear snowbank is seen in patients with sarcoidosis, Lyme disease, and intraocular inflammation associated with human T-cell lymphotropic virus type 1 (HTLV-1) infection.73,74,75,76
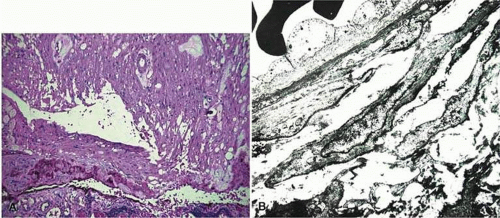 Figure 11-10. Pars planitis. A. Peripheral choroid is infiltrated by lymphocytes. (Hematoxylin and eosin ×35.) B. The “snow bank” is made up of fibroglial cells and vascular channels lined by prominent endothelial cells (×2,000). |
The ciliary body is involved in other inflammatory disorders that affect the iris, choroid, and lens. These include sarcoidosis, which appears as noncaseating granuloma, phacoantigenic endophthalmitis showing granulomatous inflammation, and diffuse granulomas seen in association with sympathetic ophthalmia.77,78,79,80,81 Infectious changes can be seen with viral infections such as varicella-zoster virus (VZV) and other herpes viruses, bacterial infections including acid-fast organisms, and various fungal and parasitic infections. VZV infection can cause necrosis of the ciliary body and may show granulomatous inflammation.82 The bacterial infections result in both suppurative and nonsuppurative inflammations as well as granulomatous infiltration. The latter is typically seen with tuberculosis.83
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


