Pathology of the Lens
Marilyn C. Kincaid
The lens is unique in that it is a closed structure composed of epithelium with its basement membrane on the outside. It is transparent and has no innervation or blood supply.
During the first few weeks of embryonic life, surface ectoderm overlying the optic cup invaginates and pinches off to form a sphere of cells surrounded by basement membrane. The surface ectoderm later forms the eyelid skin, conjunctival epithelium, and corneal epithelium. The posterior cells of the lens vesicle elongate, losing their nuclei and forming the lens fibers, while the anterior cells remain as low cuboidal epithelium a single layer thick. Lens fibers are termed as such because they are long and slender; there is no true connective tissue in the lens.
The lens epithelium keeps replicating throughout life at the lens equator, reflecting its surface ectodermal origin. Because the cells cannot be desquamated, the lens very gradually becomes larger anteroposteriorly throughout life.
CONGENITAL ANOMALIES
Abnormalities of the lens are seen in many ocular and systemic congenital malformations and diseases. Congenital anomalies can also be isolated findings, and can be hereditary or sporadic.
LENS COLOBOMA
The lens has a natural tendency to assume a more spherical shape. This phenomenon accounts for accommodation, when the circular muscle of the ciliary body contracts, allowing the zonules to relax. This tendency probably also explains lens coloboma, in which the lens zonules are missing in the area of a ciliary body coloboma and the lens appears notched in that area. Because there are no zonules in the area of the coloboma, the lens takes on its more natural spherical shape, forming a notch in this area.
CONGENITAL CYSTIC EYE
A congenital cystic eye develops when the optic vesicle fails to invaginate, an event that normally takes place at 4 weeks’ gestation. When this happens, the expected contact between the optic cup and the surface ectoderm does not occur. Thus, a congenital cystic eye contains no surface ectodermal component, including the lens.1
ANOPHTHALMIA
Anophthalmia can be primary or secondary, involving central nervous system malformations, or it can be degenerative. In primary and secondary anophthalmia, no optic vesicle is formed and thus the lens is not induced.2 The term degenerative anophthalmia implies the formation and subsequent loss of the optic vesicle. Therefore, in degenerative anophthalmia, the optic vesicle may have existed long enough to induce lens and cornea formation, so remnants of these structures may be found within the orbit.3
The lens may be unformed or resorbed in a number of other conditions. In one bilateral case of congenital absence of the pupils, both lenses were also missing, replaced by fibrous, calcific tissue.4
SYNOPHTHALMIA
Synophthalmia and cyclopia result when the brain does not form two hemispheres. This is called holoprosencephaly, and it leads to failure of the eyes and face to form properly. The eyes are partially fused in synophthalmia and completely fused in cyclopia. In synophthalmia, the more anterior structures are duplicated, and can appear quite normal, but the eye is fused posteriorly and is more disorganized. Cyclopia is extremely rare and results in a single, relatively well-formed eye, since the surface ectodermal structures are properly induced by the optic cup.5 An apparently similar anomaly is the formation of two eyes in a single orbit; however, this is more likely a localized anomaly, in which a fold was formed in the neuroectoderm of the primary optic vesicle.6
MITTENDORF’S DOT
Although not visually significant, Mittendorf’s dot, a small remnant of the fetal hyaloid vascular system, may be found on the posterior surface of the lens. Typically it is just inferonasal to the posterior pole.7
MESENCHYMAL DYSGENESIS
Much of the tissue of the anterior segment of the eye is derived from the neural crest.8 In one form of Peters’ anomaly, the lens fails to separate from the cornea and to undergo normal inward migration. Clinical findings in such cases are a central corneal leukoma, iris strands attached to the corneal stroma, and anterior cataract. In other forms, the lens separates and forms normally. Peters’ anomaly can be an isolated, uniocular finding or part of other ocular and systemic malformations; thus, it appears to be a morphologic abnormality rather than a specific diagnosis. It can be inherited as an autosomal-dominant8 or -recessive trait. Some cases have been associated with chromosomal abnormalities.9 Histologic findings in Peters’ anomaly are disorganized central corneal stroma and a gap in Descemet’s membrane. There is typically fibrous tissue between that membrane and the adherent lens.10 In one patient with Peters’ anomaly who also had persistent hyperplastic primary vitreous, only remnants of the lens capsule remained.11 Even if the lens is not adherent to the cornea, there may be a cortical cataract.8
LENTICONUS
The lens can assume a focally conical shape either anteriorly or posteriorly. Anterior lenticonus apparently occurs only as part of Alport’s syndrome, and it is clinically evident as a central nipple-like lenticular protrusion and myopia. The capsule is unusually fragile, and this finding correlates with irregular capsular dehiscences noted on electron microscopic examination. The molecular defect apparently involves a noncollagenous, globular protein domain of type IV collagen.12 Posterior lenticonus can be unilateral or bilateral, and the red reflex looks like an oil droplet. The cause is obscure but may involve either traction or a focal weakness in the capsule. Secondary subcapsular or cortical cataract can occur.13
ZONULAR DEFECTS
Zonules are composed of a glycoprotein with a high amount of cystine; biochemically they are part of an elastic microfibrillar system.14 Lens dislocation and subluxation are components of Marfan’s syndrome, homocystinuria, and Weill-Marchesani syndrome.
Marfan’s syndrome is a connective tissue disorder characterized by an upward subluxation of the lens. The zonular and capsular fibers have been shown by electron microscopy to be large and granular, with a loss of their normal parallel orientation.15
Homocystinuria is a disorder of homocysteine metabolism. The lenses tend to be dislocated by age 10. The zonular fibers are decreased on the lens surface and recoiled onto the surface of the ciliary body. The nonpigmented ciliary epithelium is atrophic.16 Because the zonular protein has a high sulfur content, disorders of sulfated amino acid metabolism would be expected to cause zonular abnormalities.14
Weill-Marchesani syndrome is a musculoskeletal disorder of unknown cause. The lenses are small and spherical, and affected patients are highly myopic. Lens dislocation tends to occur in a downward direction. The molecular defect is not known.17
Ectopia lentis et pupillae is an autosomalrecessive disorder characterized by lens and pupillary displacement, axial myopia, retinal detachment, cataract, persistent pupillary membranes, iridohyaloid adhesions, and megalocornea. Thecause is not known, and the ocular findings can vary not only among different families, but also between the two eyes of an affected person. Zonules are abnormal and missing, and this disease may also represent an abnormality in their biosynthesis.18
CONGENITAL CATARACT
Congenital cataract is one of the most common significant ocular disorders in infants. Cataracts can occur as an isolated defect, and may be sporadic or familial in any of the Mendelian patterns. Gene loci on chromosomes 1, 2, and 16 have all been implicated in the autosomal-dominant variety. Cataracts can also be autosomal recessive or associated with chromosomal translocations.19
One type of autosomal-dominant cataract is termed zonular pulverulent cataract, because these cataracts are powdery in appearance19 and because they reflect a limited disruption seen as opaque fibers in only one zone of the lens.20 They are usually symmetric but can vary in appearance not only among different members of the same pedigree, but also between the two eyes of an affected person. They can range from being visually insignificant to causing major disability.19
Zonular cataracts are termed as such because they affect only one zone of lens fibers and are nonprogressive, but because the lens continues to form new fibers throughout life, the involved lens fibers gradually are pushed more centrally. Acquired types occur because of trauma or metabolic disorders. On histologic examination, the fibers are seen to have been replaced by disorganized, variably sized globules.20
Galactose cataracts are seen in disorders of galactose metabolism. Galactose, a monosaccharide, is combined with glucose to form the disaccharide lactose. Affected persons therefore cannot metabolize milk products properly. There are three enzymes21 in the metabolic pathway that can be defective; the genes are located on different chromosomes, and defects are inherited as autosomalrecessive traits. The sugar alcohol galactitol accumulates in the lens, resulting in water imbibition and cataract. Early in the course of the disease, this cortical opacity may be reversible.21
Myotonic dystrophy is a muscle disorder characterized by frontal balding and distinctive facies. Affected patients have difficulty relaxing contracted muscles. The characteristic cataract is in the deep subcapsular region and consists of fine, iridescent, bluish particles. Ultrastructurally they correspond to vacuoles with multilaminar membranes. These laminations apparently are responsible for the iridescent color.22
In Lowe’s syndrome, there are ocular abnormalities along with mental retardation and renal tubular acidosis.23 Lowe’s syndrome is inherited as an X-linked recessive trait, and female carriers may show lens opacities.24 Tripathi and coworkers25 have suggested that the primary lenticular defect resides in the lens cells.
Soemmering’s ring cataract appears to be due to defectively formed primary posterior lens fibers that subsequently degenerate, giving rise to a centrally flattened or ring-shaped cataract. There is no demarcation between nuclear and cortical fibers, and lens nuclei are retained. Other lens changes are nonspecific. There can be focal excrescences of the lens capsule,25 which have also been described in Down’s syndrome (trisomy 21).26 Down’s syndrome patients often have cataracts, but these tend to be nonspecific and to occur in older Down’s syndrome patients. In one series, the youngest patient with clinically and histologically evident opacities was 15 years of age.26
CAPSULE
The capsule is, at least in the paracentral region, the thickest basement membrane of the body. Like basement membranes elsewhere, the lens capsule can stretch, so the shape of the lens can be distorted, within limits, without capsular rupture. The intact lens can prolapse through corneal defects, and this is often how the lens is lost when the eye is traumatically ruptured, even if the corneal defect is relatively small.
TRUE EXFOLIATION
True exfoliation of the lens capsule is a condition in which the anterior aspect of the capsule splits. Clinical findings are a thin, clear, colorless membrane floating in the pupillary axis. It was first described in glass blowers and was believed to be a consequence of the considerable infrared radiation to which they were exposed. It has also been described in blacksmiths and steelworkers as well as in other persons who have had similar infrared exposure.27
It is possible, however, for patients to develop true exfoliation without having an apparent history of infrared radiation exposure. True exfoliation secondary to severe iridocyclitis or occurring as a sequela of trauma has been suggested.28 In some cases, the cause is obscure, but it may be a change associated with aging. In the largest reported series of 11 eyes in seven patients, the youngest patient was 79. All were hyperopic.27
Histologic examination shows an unequal splitting of the anterior capsule (Fig. 1). Electron microscopic examination shows that the lens capsule is laminated and the anterior layer or layers are split free.29 The free portion is quite thin, and it can be rolled or folded over.27
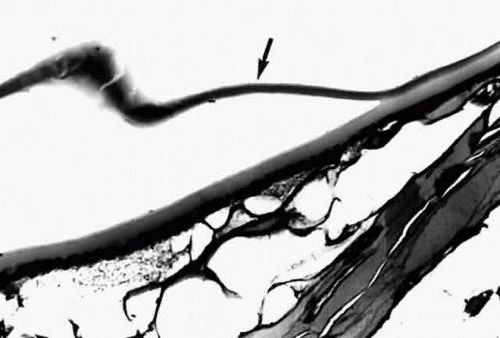 Fig. 1. True exfoliation of the lens capsule. The anterior lens capsule is split and floats away from the rest of the lens (arrow). The subepithelial disruption is artifact. (H & E, magnification × 234; Courtesy of Dr. W.R. Green, Baltimore, MD) |
PSEUDOEXFOLIATION SYNDROME
Pseudoexfoliation of the lens is much more common than true exfoliation. The term is actually a misnomer, as it has become clear that the origin of the pseudoexfoliation material is not the lens capsule. It is more prevalent in Scandinavian countries and is associated with secondary open-angle glaucoma.30
Clinically, in its classic manifestation, pseudoexfoliation syndrome is manifest as a whitish, fluffy deposit on the lens surface and at the pupillary margin. Typically there is a circular clear zone in the midperiphery of the lens, where the iris comes in contact with it (Fig. 2). This syndrome is important to recognize because a type of secondary open-angle glaucoma that can be difficult to control is associated with pseudoexfoliation in more than 20% of eyes.31 Other associated changes include poor dilatation of the iris, melanin dispersion from degeneration of the iris pigment epithelium, and iris stromal atrophy, which are visible clinically as transillumination defects.32
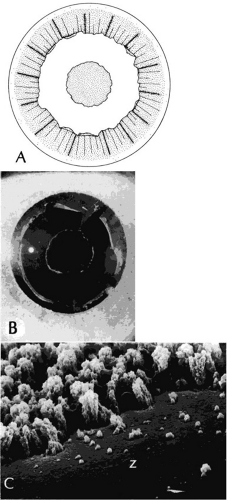 Fig. 2. Pseudoexfoliation syndrome. A and B. Central disc surrounded by a relatively clear zone, surrounded in turn by a peripheral granular area. C. Scanning electron micrograph shows relatively clear zone (Z) surrounded by the central edge of the peripheral granular area. (B, courtesy of Dr. G. Naumann, SEI 73-985; C, courtesy of Dr. R.D. Eagle, Jr;) magnification × 900) |
Because of its appearance, it was natural to assume that the exfoliation material was derived from the lens capsule. In some eyes, however, it was observed to persist or to develop long after intracapsular lens extraction. The stimulus for its deposition is likewise unknown. Subsequently, the material has been found in the conjunctiva,33 iris,32 and visceral organs.31 Thus, pseudoexfoliation syndrome appears to be a systemic disease, although there are no clinical symptoms apart from those in the eye. The syndrome has been described as typically unilateral, but it now appears that it is more frequently bilateral but asymmetric. Sometimes, too, eyes with uncontrolled open-angle glaucoma and other signs of exfoliation, such as poorly reactive pupils, but with no evidence of intraocular exfoliation material, can be shown to have the syndrome by conjunctival biopsy.33
Histologic examination shows exfoliation deposits appearing as short strands of eosinophilic material, which are found on the surface of the lens and zonules and within and adjacent to the ciliary body and iris (Color Plate 1A). On electron microscopic examination, these strands are found to be thick, straight, and densely osmiophilic, with associated thin microfibrils.33
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


