Pathology of Conjunctiva
Tatyana Milman
The conjunctiva is a mucous membrane that plays a critical role in maintaining ocular health by forming a smooth, flexible, and protective sac covering the pericorneal surface of the eye.1 An intact conjunctiva forms a barrier to entrance of infectious organisms and provides immune surveillance and immunoreactivity for antigenic stimuli. In addition, the conjunctiva secretes the mucin component of the tear film, thus aiding in even tear distribution over the surface of the eye. Conjunctival vessels provide nourishment to the avascular cornea. The purpose of this chapter is to provide an overview of conjunctival anatomy and histology, congenital anomalies, inflammatory and infectious conditions, degenerations and deposits, manifestations of systemic disease, trauma, and tumors and pseudotumors with an emphasis on pathology. The detailed information on clinical manifestations and management of these conditions can be found elsewhere in the text.
Anatomy and Histology
Similar to the mucous membranes elsewhere in the body, the conjunctival surface is composed of nonkeratinized stratified squamous, cuboidal, and columnar epithelium with mucin-producing goblet cells. Occasional dendritic melanocytes can be observed in the basal epithelium, whereas Langerhans’ cells (dendritic antigen presenting cells) are scattered throughout more superficial epithelial layers. The conjunctival epithelium rests on a loose fibrovascular connective tissue called substantia propria. The substantia propria contains a meshwork of collagen and elastic fibers, fibroblasts, blood vessels, lymphatics, peripheral nerves, and inflammatory cells (mostly lymphocytes, plasma cells, and occasional mast cells) (Fig. 1).
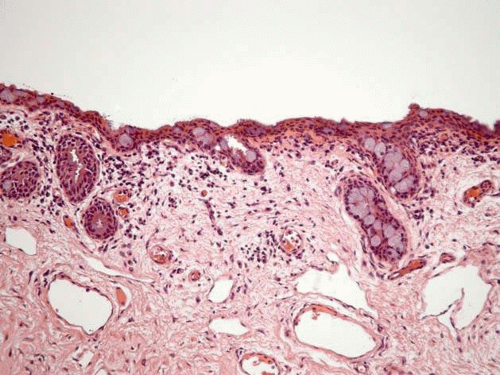 Figure 1. Normal conjunctiva. Stratified squamous nonkeratinized epithelium with gray-blue goblet cells rests on substantia propria. Focal infoldings of the epithelium, pseudoglands of Henle, are present. (Stain, H&E) |
The conjunctiva can be broadly subdivided into the following regions:
Bulbar: The bulbar conjunctiva extends from the corneal limbus to the fornices. The limbal conjunctival epithelium resembles corneal in morphology and lacks abundant goblet cells. Nasally, the bulbar conjunctiva contains a crescent-shaped fold, plica semilunaris, which is rich in goblet cells (Fig. 2), The caruncle (a small piece of flesh, lat.) is located nasal to the plica. This specialized region of conjunctiva is also rich in goblet cells and may contain hairs with associated glands, lacrimal gland tissue, lobules of fat, occasional smooth muscle fibers, and rarely cartilage. The epithelium of the caruncle may be keratinized (Fig. 3).
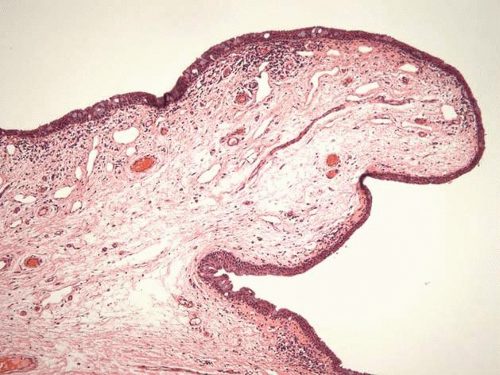
Figure 2. Plica semilunaris. (Stain, H&E)
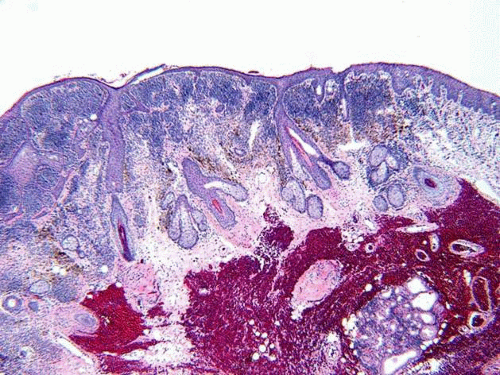
Figure 3. Caruncle is lined by stratified squamous keratinized epithelium on the left and nonkeratinized epithelium on the right. Dermal appendages are observed within the stroma, surrounded by subepithelial nevus nests. (Stain, H&E)
Tarsal: The tarsal (palpebral) conjunctiva extends from the mucocutaneous junction at the eyelid margin to the edge of the tarsal plate, and is firmly adherent to the tarsus. Tarsal conjunctival epithelium is more cuboidal in morphology and is rich in goblet cells. It forms invaginations called pseudoglands of Henle (Fig. 4).
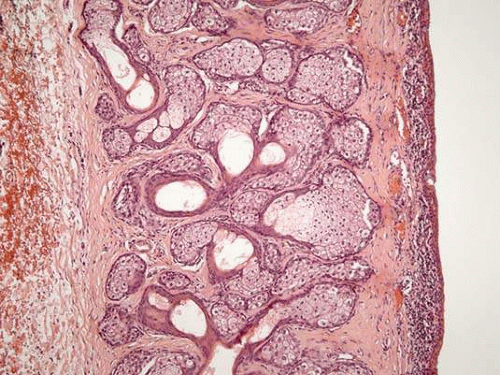
Figure 4. Tarsal conjunctiva. (Stain, H&E)
Forniceal: Forniceal conjunctiva bridges the bulbar and tarsal conjunctiva, and is a cul-de-sac region that houses the ducts of the main and accessory lacrimal glands. Nonkeratinized stratified columnar forniceal conjunctival epithelium is folded into numerous pseudoglands of Henle and is rich in goblet cells. The substantia propria of the forniceal conjunctiva adheres loosely to the underlying tissues (Fig. 5).1,2,3
Figure 5. Forniceal conjunctiva. (Stain, H&E)
Congenital and Developmental Anomalies
Various choristomatous tumors can involve conjunctiva. Choristoma is defined as presence of normal tissue elements at atypical location. Some of these lesions are discussed subsequently.
Solid Limbal (Corneal, Epibulbar) Dermoid
Limbal dermoid presents as a whitish round mass typically in inferotemporal limbal area (Fig. 6A). Some cases have been mapped to chromosome X, and display X-linked recessive inheritance.4 Limbal dermoids may be associated with Goldenhar syndrome (oculoauriculovertebral dysplasia). This syndrome is characterized by eyelid colobomas, epibulbar dermoids, accessory auricular appendages, aural fistulas, deafness, hemifacial hypoplasia of the soft and bony tissues, and vertebral anomalies. Although most cases of Goldenhar syndrome are sporadic, some have autosomal dominant inheritance.2,5,6
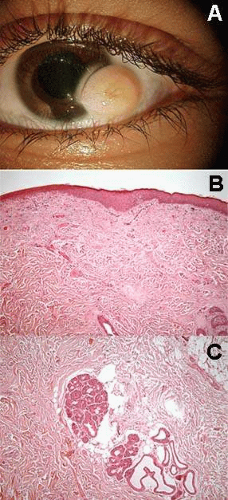 Figure 6. Solid limbal dermoid. A. White mass with surface hair shafts at inferotemporal limbus. B. The lesion is lined by stratified squamous keratinized epithelium, resembling epidermis. Coarse collagen bundles, resembling dermis are observed in the underlying stroma. Hair follicles and associated glands are also observed. C. Apocrine glands within the stroma of the lesion. (Stain, H&E) |
Histopathologically, solid limbal dermoids are composed of proliferation of choristomatous tissue not normally found in the limbal area, such as keratinized epithelium, dermal appendages, fat, smooth and striated muscle, lacrimal gland, cartilage, brain, teeth, and bone (Fig. 6B).2 When multiple tissues contribute to the lesion, it is called a complex choristoma (Fig. 7). Complex choristomas are a feature of linear nevus sebaceous syndrome (organoid nevus syndrome), a rare sporadic condition characterized by a cutaneous sebaceous nevus, seizures, mental retardation, and ocular abnormalities (Fig. 8).7,8
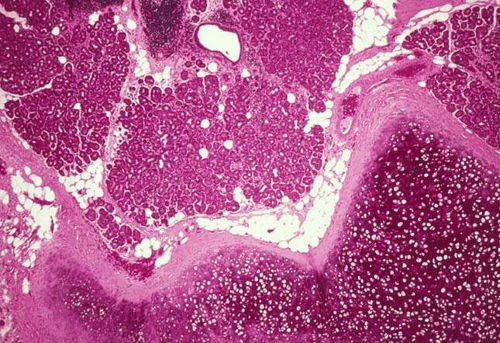 Figure 7. Complex choristoma. The lesion demonstrates the presence of various mature tissues within the lesion, including cartilage and lacrimal gland. (Stain, H&E) (Courtesy of Dr. Ralph C. Eagle, Jr.). |
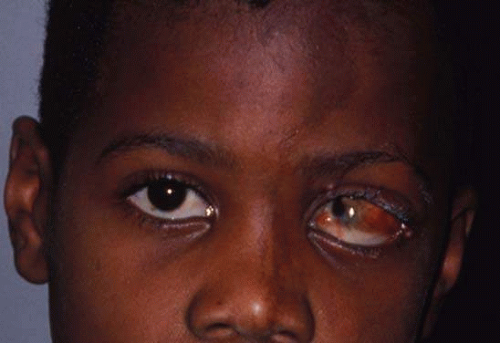 Figure 8. Epibulbar complex choristoma. A child with linear nevus sebaceous syndrome demonstrates a characteristic skin lesion on the forehead and nose and a left epibulbar lesion, histopathologically shown to be a complex choristoma. (Courtesy of Dr. Jerry A. Shields). |
Dermolipoma
Dermolipoma or lipodermoid presents as bilateral yellowish-white congenital mass on the superotemporal epibulbar surface. It is similar to solid limbal dermoid, but has a predominance of fatty tissue (Fig. 9). Dermolipomas have been associated with Goldenhar syndrome.2
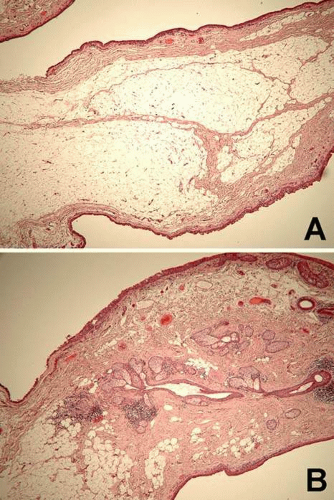 Figure 9. Dermolipoma. A. Dermolipoma is similar to solid epibulbar dermoid histopathologically, but demonstrates an abundance of adipose tissue. B. Coarse collagen bundles and dermal appendages are also present. (Stain, H&E) |
Ectopic Lacrimal Gland
Ectopic lacrimal gland is relatively common in the anterior uvea, but can also involve the substantia propria of the conjunctiva. This tumor appears as a pinkish, nodular mass (Fig. 10A). It consists of normal lacrimal gland tissue that may be associated with other choristomatous tissues, such as muscle, nerve, cartilage, and dermal appendages (Fig. 10B).9
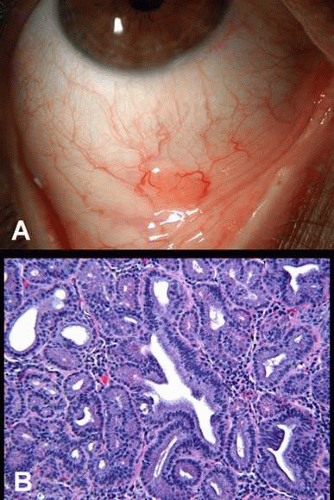 Figure 10. Lacrimal gland choristoma (ectopic lacrimal gland). A. Orange vascular mass in the inferior fornix, simulating a lymphoid tumor. B. Histopathology shows lobules of mature lacrimal gland tissue. (Stain, H&E) |
Inflammation and Infection
A variety of infectious agents, toxins, and allergens can trigger inflammation of the conjunctiva, or conjunctivitis. Conjunctivitis can be subdivided into several broad categories, such as acute and chronic, papillary and follicular, infectious and noninfectious, membranous and pseudomembranous, granulomatous, and cicatrizing.
Acute and Chronic Conjunctivitis
Clinically, acute conjunctivitis presents with a rapid onset of redness, irritation, chemosis, and copious discharge, whereas chronic conjunctivitis has a more insidious, indolent course. Histopathologically, acute conjunctivitis is characterized by predominantly acute inflammatory infiltrate, that is, neutrophils, eosinophils, and mast cells (Fig. 11), whereas in chronic conjunctivitis the inflammatory response is composed mostly of chronic inflammatory cells, namely lymphocytes and plasma cells (Fig. 12A). Additional histopathologic manifestations of chronic conjunctivitis include hyperplasia of the epithelium and goblet cells.2
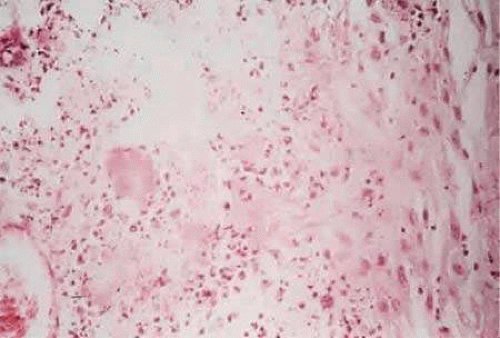 Figure 11. Acute conjunctivitis: Numerous polymorphonuclear leukocytes are scattered throughout the substantia propria of the conjunctiva |
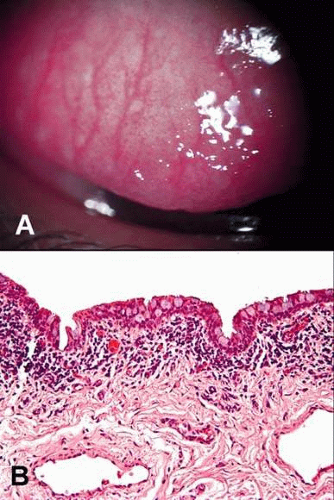 Figure 12. Papillary conjunctivitis. A. Clinical photograph shows conjunctival mounds with central vascular cores, consistent with papillae. B. Histopathology of the papilla demonstrates a central vascular core surrounded by chronic inflammatory cells. (Stain, H&E) |
Papillary and Follicular Conjunctivitis
Conjunctivitis can be also described as papillary or follicular, both clinically and histopathologically. In papillary conjunctivitis the conjunctival epithelium is elevated by an acute or chronic inflammatory infiltrate which surrounds a central fibrovascular core. The connective tissue septa surround each papilla, thus limiting the extent of inflammatory infiltrate and edema, resulting in a typical clinical appearance of an elevated polygonal mound with a central red dot (dilated capillary viewed end-on) (Fig. 12).
Papillary conjunctivitis is a nonspecific response to a noxious agent and can be a part of inflammatory reaction to any infectious organism, allergen, toxin, or trauma. Giant papillae (greater than 0.3 mm in diameter), however, are much more specific, and may be observed in vernal or giant papillary conjunctivitis (see below).
Follicles appear clinically as small, dome-shaped, pale nodules surrounded by blood vessels. Histopathologically, the conjunctival epithelium is elevated by a discrete collection of inflammatory cells (typically lymphocytes and plasma cells), occasionally forming a germinal center (Fig. 13). Follicular conjunctivitis is more specific then papillary. Follicles are a hallmark of viral conjunctivitis. They can also be observed in parasitic infections (pediculosis), bacterial infections (chlamydiae), and in toxic reaction (topical medications).1
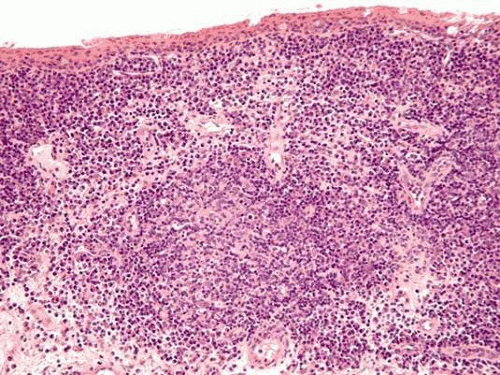 Figure 13. Follicular conjunctivitis. Conjunctival follicle appears as a round collection of inflammatory cells in a germinal center arrangement, surrounded by blood vessels. (Stain, H&E) |
Infectious Conjunctivitis
I. Bacterial Conjunctivitis
Bacterial conjunctivitis is more common in children then the viral form, whereas the opposite is true for adult population. Bacterial conjunctivitis presents classically bilaterally with papillary reaction and yellow-white purulent discharge. Several presentations and etiologic agents of bacterial conjunctivitis are discussed subsequently.
A. Hyperacute purulent conjunctivitis
Hyperacute conjunctivitis presents with the explosive onset (<24 hours) of severe purulent discharge, marked conjunctival hyperemia, chemosis, and papillary reaction. The most common etiology is N. gonorrhoeae. Gonococcal conjunctivitis has a high association with keratitis and ulceration, which may rapidly progress to perforation. Up to one third of the patients may have concurrent chlamydial venereal disease. Conjunctival gram-stained smears and cultures help establish the diagnosis. Although topical antibiotics and copious irrigation are part of the treatment, systemic treatment for gonorrhea and chlamydia is imperative.10
B. Acute purulent conjunctivitis
Acute purulent conjunctivitis presents with an acute onset (hours to days) of purulent discharge, conjunctival hyperemia, and papillary reaction which are less dramatic as compared to hyperacute conjunctivitis. The most common pathogens include Streptococcus pneumoniae, Staphylococcus aureus, Hemophilis influenzae, and Moraxella catarrhalis. The infection is usually self-limited, and is often treated with empiric antibiotics. The treatment is controversial, however, since some studies have found no increased benefit of topical antibiotics as compared to supportive care, unless the treatment was implemented early at the onset of culture-positive bacterial conjunctivitis. Conjunctival cultures are not always indicated for acute purulent conjunctivitis, however. The decision to culture may be influenced by lack of resolution of symptoms despite empiric antibiotics, by severe presentation that may be difficult to distinguish from hyperacute conjunctivitis, and by an immunocompromised state of a host.(10,11)
C. Chlamydial conjunctivitis
Chlamydia trachomatis is the microorganism that causes trachoma (serotypes A-C), neonatal and adult inclusion conjunctivitis (serotypes D-K), and lymphogranuloma venereum (serotypes L1-L3).12,13,14,15 The agent is an atypical microorganism that resembles bacteria in the sense of sensitivity to antibiotics but, like a virus, is an obligate intracellular organism. The intracellular reproduction of chlamydia produces the elementary bodies and initial bodies. The cytoplasmic inclusion body, known as an elementary body, is a small particle, 200 to 350 nm in diameter. The elementary body, after enlargement (up to 1 μm), is known as an initial body. The initial bodies divide by binary fission, multiply, and reorganize into numerous small highly infectious elementary bodies that are released from the cell. Histologically, large basophilic initial bodies can be seen to cap the nucleus of infected cells, and the accumulation of numerous initial bodies is identified intracytoplasmically by Giemsa-stained cells scraped from the surface of the infected epithelium (Fig. 14).16 Cell culture isolation and PCR are widely used methods for diagnosis of Chlamydia trachomatis infection.
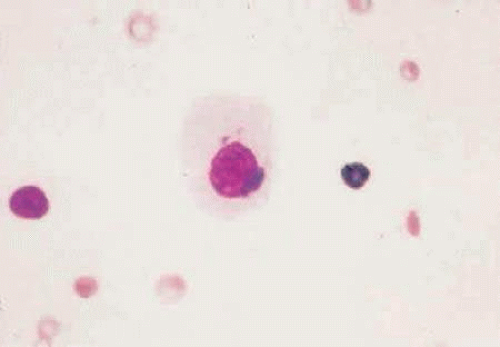 Figure 14. Photomicrograph shows large basophilic initial bodies capping the nucleus of cells infected with Chlamydia trachomatis. |
Trachoma
Trachoma is caused by C. trachomatis and is spread from eye to eye by transfer of ocular discharge. Trachoma is one of the world’s leading causes of blindness and primarily affects the conjunctiva and corneal epithelium. Clinically, one sees a mixed follicular and papillary reaction in the early phases, but as cicatrization progresses, the conjunctiva becomes white and scarred. A linear scar, known as Arlt’s line, may extend horizontally across the palpebral conjunctiva and is considered a hallmark of previous trachoma infection. Similarly, necrosis and involution of limbal follicles produces characteristic depressions known as Herbert’s pits. The World Health Organization (WHO) has introduced a simple severity grading system for trachoma based on the extent of conjunctival inflammation, tarsal conjunctival scarring, trichiasis, and corneal opacification.
The histology of trachoma is described in MacCallan’s four stages. Early (Stage I), there is epithelial hyperplasia with lymphocytic infiltrates in the basal epithelium and substantia propria. The conjunctival epithelial cells contain intracellular microcolonies of elementary bodies and large basophilic initial bodies, which form the cytoplasmic inclusion bodies of Halberstaedter and Prowazek. Florid inflammation with follicle formation are the hallmark of the next stage (Stage II), followed by the formation and predominance of papillae. Large macrophages with phagocytosed debris (Leber cells) appear in conjunctival substantia propria. Finally, goblet cells are lost, and there is epidermalization and keratinization with scar formation (cicatrization, Stage III) in the substantia propria. Stage IV marks the arrest of the disease.2,17
Active trachoma is treated with topical and systemic antibiotics. Family contacts of the patients should be screened and treated if necessary.
Inclusion Conjunctivitis
Inclusion conjunctivitis is a sexually transmitted disease, and presents as an acute purulent infection in newborns or as a subacute/chronic follicular conjunctivitis in adults.13,14 Follicles in the upper tarsal and bulbar conjunctiva and semilunar fold are a helpful diagnostic sign in adult chlamydial inclusion conjunctivitis.
The diagnosis is confirmed by examination of Giemsa-stained slides prepared from conjunctival scrapings (which demonstrate epithelial cytoplasmic inclusion bodies indistinguishable from those of trachoma), cell culture isolation, and PCR. Systemic and topical antibiotics are recommended for both neonatal and adult chlamydial conjunctivitis. In addition, the patients should be evaluated for other sexually transmitted diseases before antibiotic treatment is started. Sexual partners should be concomitantly treated to prevent reinfection.18
II. Viral Conjunctivitis
Viral conjunctivitis likely accounts for most cases of infectious conjunctivitis in adults. Viral conjunctivitis classically presents with follicular reaction, watery discharge, and regional lymphadenopathy. Corneal involvement can occur, and the pattern of involvement may aid in establishing the diagnosis. Herpes viruses (HSV, VZV, EBV), adenoviruses, poxviruses (molluscum contagiosum), and RNA viruses (measles, mumps, rubella) are some of the pathogens that have the potential to cause conjunctivitis. Immunodiagnostic methods and cytology, culture, and PCR of conjunctival scrapings can be useful diagnostic tests in selected cases. Treatment is guided by the etiologic agent of viral conjunctivitis.
Noninfectious Conjunctivitis
Noninfectious etiologies of conjunctivitis include radiation, burn, and chemical injuries, allergic and immunologic disorders, and neoplastic processes. Selected conditions are discussed subsequently.
I. Radiation Conjunctivitis
Super voltage orbital radiation therapy, including proton beam radiation therapy and plaque radiation therapy, is used widely for treatment of ocular tumors. Immediately after radiation exposure, the conjunctiva becomes hyperemic and edematous. The surface epithelium also becomes edematous and epithelial cells are sloughed. In a period of weeks to months, typical changes of epithelial metaplasia and keratinization with associated fibrosis and scarring can occur. The patients develop features of xerosis and severe dry eye.19
II. Chemical Conjunctivitis
Acid and alkali are important causative agents of chemical conjunctivitis. Acid causes coagulative necrosis of conjunctival epithelium and precipitation of proteins, thereby leading to its neutralization and limiting its penetration. Alkali, on the other hand, rapidly penetrates cell membranes, resulting in immediate swelling of the epithelium, followed by its desquamation, allowing for further penetration of the alkali. Alkali also coagulates blood vessels. Histopathologically, loss of conjunctival vessels and widespread necrosis are seen. Neutrophils may also be observed.2
III. Allergic Conjunctivitis
Allergic eye disease is common, and is more frequent in males. There are four main types of allergic eye diseases: allergic conjunctivitis, vernal keratoconjunctivitis, atopic keratoconjunctivitis, and giant papillary conjunctivitis.
A. Acute allergic conjunctivitis
Acute allergic conjunctivitis is one of the more common etiologies of a noninfectious conjunctivitis, which occurs due to the direct exposure of the ocular mucosal surfaces to environmental allergens. Seasonal allergic conjunctivitis (SAC) is a type I, IgE and mast cell-mediated hypersensitivity reaction to pollens from grasses and trees, whereas perennial allergic conjunctivitis (PAC) is a type I hypersensitivity reaction to common household allergens, such as dust mites and animal dander. Itching, mucous discharge, milky or pale pink edematous conjunctiva with papillary reaction, and eventually cicatricial changes (supepithelial fibrosis) are observed clinically. Additionally, venous congestion can cause the appearance of dark circles around the eyes called “allergic shiner,” and patients often experience rhinitis. Clinical history or family history of atopy may be present.20,21
The diagnosis is made easier by detailed clinical history. Serological tests may reveal elevated serum IgE in over 70% of the patients, whereas tear fluid IgE is present in almost all tear fluid samples. However, these findings are not specific.21 Skin testing by an allergist can pinpoint the offending allergen(s).
B. Vernal keratoconjunctivitis
Vernal keratoconjunctivitis (VKC) is a bilateral chronic mast cell-lymphocyte-mediated allergic disorder, which involves both type I and type IV hypersensitivity reactions. VKC affects predominantly prepubescent boys. Sex distribution equalizes after puberty. Strong personal and/or family history of atopy can be elicited in many patients. Seasonal peak incidence in spring is characteristic, but the condition can persist all year round, particularly in hotter tropical climates. Characteristic symptoms are intense pruritus, photophobia, blepharospasm, and copious ropy mucoid discharge.21,24
Clinical examination reveals diffuse papillary hypertrophy, typically in the upper tarsal region. Giant cobblestone-like papillae may develop in severe cases (palpebral VKC). Bulbar conjunctival hyperemia and chemosis, and formation of gelatinous-white limbal nodules (Horner-Trantas dots) are observed in limbal form of VKC (Fig. 15A), which can occur in isolation or in conjunction with palpebral VKC. Corneal changes (corneal VKC) may also be observed and include punctate epithelial erosions in the superior and central cornea and superficial shield-like ulcers, which can lead to stromal opacification and vascularization. An association with keratoconus has been reported.2,21,24
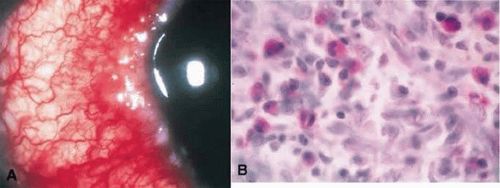 Figure 15. A. Clinical photograph shows limbal vernal conjunctivitis. B. Histopathology shows numerous inflammatory cells with a predominance of eosinophils. |
Conjunctival scrapings of the superior tarsal conjunctiva will demonstrate the presence of eosinophils and free eosinophilic granules, suggestive of degranulation. Histopathology of the superior tarsal conjunctiva shows epithelial hyperplasia and proliferation of fibrovascular connective tissue along with the infiltrate of mast cells, eosinophils, lymphocytes, plasma cells, and monocytes. Horner-Trantas dots histopathologically appear as microaggregates of degenerated eosinophils and epithelial cells (Fig. 15B).2,25
C. Atopic keratoconjunctivitis
Atopic keratoconjunctivitis (AKC) is a more serious, potentially sight threatening condition, associated with atopy. Unlike VKC, AKC is a perennial, T cell-mediated type IV immune hypersensitivity reaction, although seasonal exacerbations reflecting a compounding type I hypersensitivity response also occur. AKC can present in youth, albeit usually at an older age then VKC, and exhibits a progressive course as the patient matures. Presenting signs include bilateral atopic dermatitis of the eyelids, blepharitis, conjunctivitis, cataracts, and corneal punctate epithelial erosions, ulceration, and vascularization. Keratoconus has been also associated with AKC. In contrast to VKC, papillae in AKC are small- to medium-sized, are equally distributed on tarsal and palpebral conjunctiva, and may even show lower tarsal conjunctiva predilection. Conjunctival scarring with symblepharon formation may occur in advanced stages of the disease.21,25
Tear fluid may show increase in IgE concentration. Conjunctival cytology may reveal eosinophils, although they are not as numerous as in VKC, and are less often degranulated. Conjunctival epithelium may show an infiltrate of mast cells and eosinophils, epithelial pseudogland formation, and increased goblet cell density. Mast cells and mononuclear cell infiltration are prominent in the substantia propria. Fibroblast proliferation and fibrosis can also be observed, reflecting cicatrizing changes in late disease.21,25,26
D. Giant papillary conjunctivitis
Giant papillary conjunctivitis (GPC) has been originally described in eyes with foreign bodies (ocular prosthesis, extruded scleral buckle, and corneal foreign bodies), but has become increasingly more common with the advent of soft contact lenses.21 The etiology of GPC is not fully understood, and may involve an immune-mediated response to repeated mechanical trauma or to a type IV hypersensitivity to contact lens-related material.27
Symptoms are similar to VKC, and include itching with a mucoid ropy discharge. Foreign body sensation and intolerance to contact lens wear are also common. Clinical examination will reveal giant papillae on superior tarsal conjunctiva. Although giant papillae have been originally defined as being greater then 1 mm in diameter, it is now believed that papillae 0.3 mm or larger, in association with the other signs and symptoms of contact lens-induced conjunctivitis, are diagnostic of GPC.
Histopathology of the conjunctiva in the region of giant papillae demonstrates thickened and irregular epithelium, with prominent pseudoglands of Henle. Goblet cells may be decreased in density in the epithelium at the apices of the papillae and increased in number in interpapillary crypts. Mast cells, eosinophils, and basophils are observed both within the epithelium and in the substantia propria (Fig. 16). Abnormally elevated concentrations of immunoglobulins (IgE, IgG, and IgM, and complement components) have been found in tears of affected patients. These findings suggest a combined mechanical and immune-mediated pathophysiology of GPC.28,29
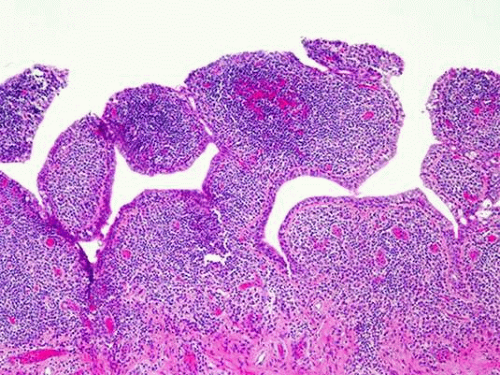 Figure 16. Giant papillary conjunctivitis. Giant papillae of a contact lens wearer with GPC. (Stain, H&E) (Courtesy of Dr. Margaret McLaughlin). |
The management of patients with allergic conjunctivitis is directed towards a specific entity, and includes allergen avoidance, climatotherapy, topical medications (antihistamines, masts cell stabilizers, steroids, cyclosporine drops), systemic medications (cyclosporine), and, occasionally, cryoablation or surgical removal of giant papillae.
IV. Immunologic Disorders of the Conjunctiva
Numerous immunologic disorders can affect conjunctiva, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis, ocular cicatricial pemphigoid, drug-induced pemphigoid, linear IgA bullous dermatosis, graft-versus-host disease, dermatitis herpetiformis, epidermolysis bullosa, lichen planus, paraneoplastic pemphigus, pemphigus vulgaris, pemphigus foliaceous, discoid lupus erythematosus, and phlyctenular conjunctivitis. Selected conditions are discussed below.
A. SJS and toxic epidermal necrolysis
Erythema multiforme major is a complex immunological syndrome, which manifests in a vesiculobullous reaction of the skin and mucous membranes. Toxic epidermal necrolysis (TEN) is the most severe form of erythema multiforme, characterized by extensive sloughing of the epidermis and mucous membranes. Autoimmune response triggered by drugs (antibiotics, anticonvulsans, salicylates) or infectious organisms has been implicated in the pathogenesis of erythema multiforme.30,31,32
The initial ocular manifestations of SJS/TEN include eyelid edema, erythema, crusting, and conjunctival hyperemia. Severe conjunctival chemosis, membrane, and pseudomembrane formation are observed less commonly. In later stages of the disease, severe dry eye and conjunctival scarring with symblepharon formation can occur. Some patients may also develop recurrent conjunctival inflammation unrelated to mechanical irritation or dryness, possibly linked to immune-complex vasculitis.30,31,32,33
Histopathology of the conjunctiva in the acute stage of SJS shows vacuolar interface mucositis, necrotizing epithelial cells, and perivascular lymphocytic infiltration. More superficial conjunctival involvement is observed in TEN, characterized by intraepithelial inflammation and bullae and extensive epithelial sloughing. In chronic disease, fibrosis and loss of goblet cells may be observed, and mononuclear inflammatory cell infiltration is present in the substantia propria (Fig. 17A). Conjunctival biopsy in patients with recurrent conjunctivitis after SJS occasionally reveals vasculitis or perivasculitis, immunoreactant deposition in vessel walls, vascular basement membrane disruption, thickening, and reduplication, and a preponderance of helper T lymphocytes, macrophages, and Langerhans’ cells. Rarely, cicatricial pemphigoid with its representative pathologic findings is observed as a sequela of SJS (see the subsequent text).30,31,32,33,34
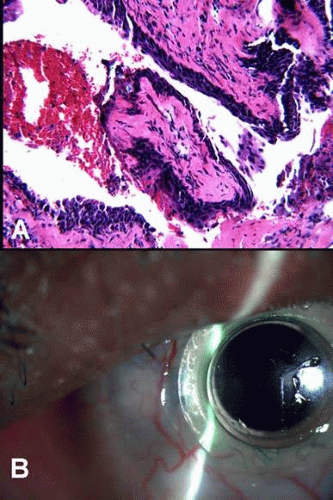 Figure 17. Stevens-Johnson’s syndrome. A. Corneal biopsy from a patient with Stevens-Johnson’s syndrome demonstrates replacement of the corneal epithelium with conjunctival-type epithelium. Focal keratinization is observed. No goblet cells are present. The epithelium rests on a fibrous pannus. (Stain, H&E) B. The patient subsequently underwent keratoprosthesis placement. |
Management of SJS/TEN is mainly supportive with ocular lubrication and surveillance for infection. Oral and topical corticosteroids are sometimes used, but their role is controversial. Symblephara can be treated by lysis and use of rings, but this is also a controversial treatment modality. Surgical correction of eyelid malposition and trichiasis, forniceal reconstruction, and keratoprosthesis for corneal complications can be attempted in the later stages of SJS/TEN (Fig. 17B).30
B. Ocular cicatricial pemphigoid
Ocular cicatricial pemphigoid (OCP) is a type II immune-mediated hypersensitivity disorder, characterized by deposition of autoantibodies to the basement membrane zone (BMZ) at the epithelial-subepithelial junction of mucous membranes (oral cavity, eye, nose, pharynx, larynx, trachea, esophagus, genitalia, and anus) and occasionally at the dermo-epidermal junction of the skin. Some of these autoantibodies are directed against the ß4 integrin subunit and laminin 5/epiligrin (IgG), whereas others are reacting with as yet uncharacterized 45 kd antigen (IgA). Binding of the antibodies to the target antigens at BMZ leads to complement activation, deposition, and inflammatory cell infiltration, manifesting clinically by subepithelial bullae formation and eventual cicatrization. Rarely, patients with OCP demonstrate circulating IgG and IgA autoantibodies and display serologic activity against classical bullous pemphigoid antigens. Environmental triggers, such as topical medications, have been occasionally implicated in pathogenesis of OCP in genetically predisposed individuals.30,35,36,37
The disease most commonly begins in the fifth or sixth decade of life and has a female predominance. The conjunctiva is involved in approximately 80% of patients with mucous membrane pemphigoid. The conjunctival lesions begin in one eye, but bilateral involvement eventually occurs. The clinical course of OCP is characterized by slow relapsing-remitting progression from chronic conjunctivitis to subepithelial fibrosis, fornix foreshortening, symblepharon and ankyloblepharon formation, corneal ulceration and opacification, and ocular surface keratinization.30,35
Histopathology of the conjunctiva in acute stage of OCP shows subepithelial bullae with predominantly subepithelial inflammatory infiltrate, composed mostly of T lymphocytes, and to a lesser extent of macrophages, dendritic cells, and neutrophils. Later in the course of disease, squamous metaplasia and loss of goblet cells are observed within the epithelium. Activation of fibroblasts leads to abnormal deposition of extracellular matrix and collagen in the substantia propria in the cicatrizing stages of OCP. Direct immunofluorescence, immunoperoxidase technique, and immunoelectron microscopy may demonstrate deposition of IgA, IgG, IgM, and C3 along the BMZ, but negative conjunctival biopsy does not exclude the diagnosis of OCP (Fig. 18).36,37,38,39
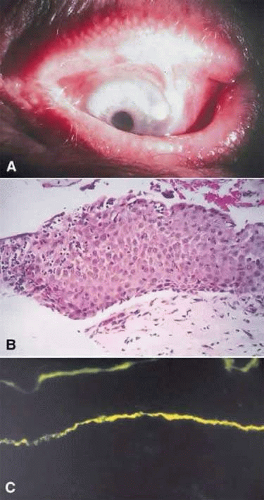 Figure 18. A. Clinical photograph shows cicatricial changes in the bulbar and palpebral conjunctiva with symblepharon in benign mucosal pemphigoid. B. Histopathology shows epithelial bullae with a slight perivascular, lymphocytic infiltrate in the substantia propria. C. Immunofluorescent stains show linear deposition of IgG and IgA along the basement membrane zone of the conjunctival epithelium. |
Medical therapy for OCP includes systemic methotrexate, cyclophosphamide, dapsone, and, possibly, intravenous immunoglobulin (IVIg). Topical corticosteroids may suppress the inflammatory response during acute exacerbations. Topical Vitamin A has been shown to reverse, to some extent, keratinization. Surgical therapies include correction of eyelid deformities, mucosal grafting for fornix reconstruction, cultivated corneal/limbal epithelial cell transplantation with amniotic membrane grafting, and keratoprosthesis.30,35
C. Linear IgA bullous disease
Linear IgA bullous disease (LABD) is an autoimmune subepithelial and subepidermal blistering disorder, which typically affects middle age adults, but has also been reported in children. Purely ocular form can occur, which is difficult do distinguish clinically and pathologically from OCP.
Histopathology is similar to OCP. Immunofluorescence demonstrates linear deposition of IgA along BMZ. Deposits of IgG/C3 may also be present. Circulating IgA antibodies to BMZ, epidermal (epithelial), and dermal (stromal) sites can occur in some patients and are helpful in establishing the diagnosis of LABD. Treatment is similar to OCP.
D. Phlyctenular keratoconjunctivitis
Phlyctenulosis is a local conjunctival and/or corneal inflammation that is believed to be caused by a type IV, cell-mediated, delayed immune hypersensitivity reaction induced by microbial antigens, such as the cell wall components of staphylococcus in developed countries or to Mycobacterium tuberculosis protein in the populations with endemic tuberculosis.40,41
Phlyctenulosis is typically a disease of the first two decades of life with a strong female predominance. Phlyctenules typically present unilaterally at or near the limbus as one or more small rounded, elevated hyperemic inflammatory nodules, accompanied by engorged vessels. They typically ulcerate centrally and spontaneously involute over a period of several weeks.40
Cultures of the eyelids and conjunctiva may be performed in cases when the clinical diagnosis is in doubt, or when phlyctenulosis does not respond to treatment. Heavy growth of S. aureus is characteristic, but negative culture does not rule out the diagnosis.
Histopathology of the phlyctenule shows an initial accumulation of plasma cells, T lymphocytes, and histiocytes in the absence of epithelial ulceration. In later stages, an infiltrate of neutrophils with secondary necrosis and ulceration occur. Organisms are usually not demonstrated within phlyctenules.40,41
Treatment consists of antibacterial/antimycobacterial and anti-inflammatory measures. Lid hygeine for staphylococcal blepharitis is also advocated to decrease the bacterial burden.
V. Neoplastic Conjunctivitis
Infiltration of the conjunctival epithelium by neoplastic processes, such as sebaceous carcinoma, can masquerade as chronic unilateral conjunctivitis. See Tumors and Pseudotumors section.
Miscellaneous Conjunctival Inflammatory/Infectious Disorders
I. Membranous and Pseudomembranous Conjunctiviitis
Membranous and pseudomembranous conjunctivitis occurs when the inflammatory discharge rich in fibrin coagulates on the conjunctival surface. A pseudomembrane lies superficially on the epithelial surface and can be peeled away without bleeding. In contrast, a true membrane incorporates conjunctival epithelium and/or granulation tissue and peels away with bleeding. The formation of pseudomembrane or a membrane often reflects the difference in the intensity of an inflammatory process, and both membranes and pseudomembranes may be present at the same time.42
The components of the exudate in membranes and pseudomembranes may point to an etiology of conjunctivitis. For example, acute membranes in immunologic and infectious disorders are composed of fibrinous exudate, neutrophils, and necrotic epithelial cells, whereas in ligneous conjunctivitis, lymphocytes, plasma cells, mast cells, and immunoglobulins predominate, with the background of extensive fibrin deposition and granulation tissue formation.42,43,44,45
Some of the etiologies of pseudomembranous and membranous conjunctivitis are listed below:
| INFECTIOUS | Bacterial conjunctivitis β-hemolytic streptococcus Neisseria gonorrhoea Neisseria meningitidis Corynebacterium diphtheria Chlamydia Viral conjunctivitis Adenovirus Herpes simplex |
| NONINFECTIOUS | Chemical burns SJS/TEN Ocular cicatricial pemphigoid and related disorders Graft-versus-host disease Ligneous conjunctivitis |
Ligneous Conjunctivitis
Ligneous conjunctivitis is a rare, usually bilateral, chronic membranous and pseudomembranous disorder, which typically presents in childhood, although rarely adults can be affected as well. The hallmark of ligneous conjunctivitis is the formation of firm, wood-like, yellowish membranes usually on the superior tarsal conjunctiva, and less frequently on bulbar and inferior tarsal conjunctiva (Fig. 19A). Other mucous membranes can be involved, including oral cavity, respiratory tract, middle ear, and genital tract. Although many reports describe spontaneous onset of disease, ligneous-like conjunctival changes may also occur after ocular surgery. Recently, patients with ligneous inflammation were found to have homozygous mutations in the type-1 plasminogen gene. Plasminogen deficiency results in defective fibrinolysis, thus manifesting in extensive fibrin deposition in inflamed or injured tissues.46,47
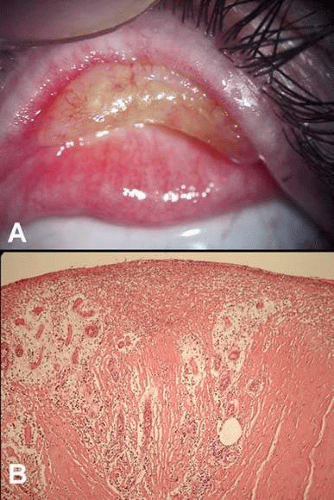 Figure 19. Ligneous conjunctivitis. A. Yellowish vascular membrane on the tarsal conjunctiva gives an appearance of “woody thickening.” B. The ligneous membrane is composed of eosinophilic fibrin deposits and granulation tissue. (Stain, H&E) |
Abundant subepithelial deposits of amorphous, acellular, eosinophilic, periodic acid-Schiff (PAS) positive material, which consists of fibrin, immunoglobulin deposit (usually IgG), and mucopolysaccharides
Granulation tissue
Inflammatory infiltrate, which is rich in T lymphocytes, although B lymphocytes, plasma cells, and mast cells can also be observed
The surface of the ligneous membrane may lack normal epithelium, and contain instead a collection of fibrin and inflammatory cells. This superficial layer may be scraped without bleeding, thus imparting a “pseudomembrane” characteristics to the superficial layer of the ligneous membrane.
Occasionally, foreign material and bacteria have been demonstrated in the ligneous membranes, but they are not believed to be responsible for the etiology of ligneous inflammation.
Ligneous conjunctivitis is usually a self-limited disorder. Serious ocular complications can result, however, including secondary infection and corneal ulceration with perforation. Treatment modalities include surgical excision of the membranes with or without adjunctive cryotherapy and amnionic membrane grafting, although recurrences are frequent. Anecdotal successes with topical corticosteroids, cyclosporine, heparin, purified plasminogen, and with intravenously administered purified plasminogen concentrate have been described.46,50,51
II. Granulomatous Conjunctivitis and Parinaud Oculoglandular Syndrome
The hallmark of granulomatous inflammation is the presence of epithelioid histiocytes and giant cells. Conjunctival granulomas can be observed in a variety of infectious and noninfectious disorders. Some of the etiologies are discussed in the subsequent text.
A. Sarcoidosis
Conjunctival sarcoidosis typically presents as discrete, yellowish, translucent nodules less then 2 mm in diameter, typically located on the inferior tarsal conjunctiva and in the inferior fornix, and less frequently on the bulbar conjunctiva.52 Occasionally, discrete, white, breadcrumb-like deposits scattered over the bulbar conjunctiva are observed in the patients with sarcoidosis.53
Histopathology of a conjunctival sarcoid nodule reveals discrete noncaseating granulomas, with central collection of epithelioid histiocytes, Langhans-type giant cells, and foreign body giant cells, surrounded by a rim of chronic mononulcear inflammatory cells (lymphocytes and plasma cells) (Fig. 20A). Proteinaceous, star-shaped (asteroid) bodies can be found in the cytoplasm of epithelioid histiocytes or foreign body giant cells (Fig. 20B). Basophilic, calcific, lamellated, birefringent Schaumann bodies are also found either within the cytoplasm of epithelioid histiocytes/giant cells or extracellularly. These bodies are not pathognomonic of sarcoidosis. White, breadcrumb-like conjunctival deposits in patients with sarcoidosis were found to consist of a collection of Schaumann bodies, surrounded by epithelioid histiocytes and giant cells.2,53
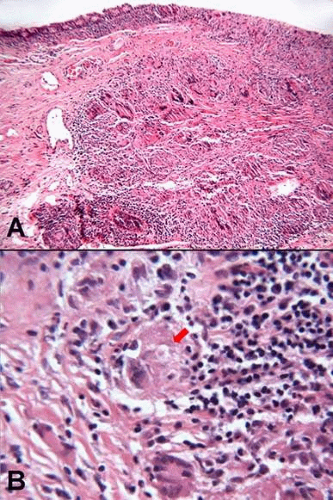 Figure 20. Conjunctival sarcoid nodule. A. Non-necrotizing granuloma in the conjunctival substantia propria in a patient with sarcoid. Langhans-type giant cells are observed. B. Eosinophilic star-shaped asteroid body is identified within one giant cell. (Stain, H&E) |
Blind biopsy of conjunctival fornix without clinically visible lesions in a patient with high clinical suspicion of sarcoidosis may demonstrate small granulomas, although the yield is low. Evaluation of serial sections from several levels may maximize the yield of biopsy.2
B. Ophthalmia nodosa
The term ophthalmia nodosa has been originally used to describe a granulomatous, nodular reaction of the conjunctiva and iris to caterpillar hairs. Later, the term has been expanded to describe ocular reactions to vegetable or insect hairs, such as tarantula hairs.54 (54)
Five types of ocular reaction to caterpillar hair have been described:54
Type 1 – the acute, toxic reaction after initial exposure to hairs, manifested by chemosis and epiphora
Type 2 – chronic mechanical keratoconjunctivitis caused by hairs lodged in the bulbar conjunciva, occasionally resulting in linear corneal abrasions
Type 3 – granulomatous phase in which the hairs incite intense foreign body granulomatous reaction
Type 4 – intense iritis caused by hairs migrating into the anterior chamber
Type 5 – vitreoretinal involvement as the hairs gain access into the posterior chamber
Histopathology of the conjunctival nodule in type 3 reaction reveals foreign material (hairs), surrounded by foreign body granulomatous reaction.
Treatment depends on the level of involvement, and includes irrigation and removal of hairs in type 1 and 2 reactions, excision of granulomatous nodules in type 3 reaction, surgical removal or laser photocoagulation of intraocular hairs, topical/ intraocular steroids, and prophylactic antibiotics.54
C. Conjunctival granulomas, associated with Splendore-Hoeppli phenomenon
Conjunctival granulomas, associated with Splendore-Hoeppli phenomenon Conjunctival granulomas may occasionally display deposition of amorphous eosinophilic material, surrounded by epithelioid histiocytes, multinucleated giant cells, lymphocytes, and eosinophils. The nature of the material has been reported as either antigen-antibody complexes, rich in immunoglobulins (IgG, IgM, IgA) and complement (C3), or as the major basic protein of eosinophils. It is hypothesized that temporal relationship between the formation of immune complexes, compliment fixation, chemotaxis, and eventual influx of eosinophils are responsible for varying composition of Splendore-Hoeppli material.55,56,57
D. Synthetic fiber granuloma
Synthetic fiber granuloma (“teddy bear granuloma”) of the conjunctiva results from an inflammatory reaction to synthetic fibers that become implanted in the conjunctival fornices. The lesion classically presents unilaterally in the inferior fornix of a child, who sleeps with a stuffed animal or a blanket made of synthetic material.58,59
Histopathology is diagnostic, and reveals numerous acellular synthetic fibers that are fairly uniform in diameter, ranging from 17 to 29 μm. The fibers lack central hollow core, do not display PAS-positivity, show parallel chatter marks from microtome blade, demonstrate dark granules on their surface (de-lustering agent), and exhibit a markedly pronounced birefringence with polarized light due to their highly organized chemical composition. These findings are helpful to distinguish the synthetic fibers from human hairs, ophthalmia nodosa, and other inorganic material. The synthetic fibers may be surrounded by Splendore-Hoeppli phenomenon. Foreign body granulomatous reaction to the synthetic material is prominent in the lesion (Fig. 21).58,59
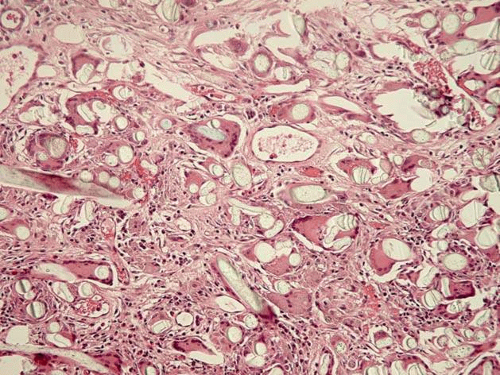 Figure 21. Synthetic fiber granuloma (“Teddy bear granuloma”). Numerous synthetic fibers are surrounded by an intense foreign body reaction. (Stain, H&E) |
The treatment involves surgical removal of foreign material and associated inflammatory reaction.
E. Actinic granuloma of the conjunctiva
It is hypothesized that actinic granuloma of the conjunctiva represents a cell-mediated immune response to actinically altered elastotic fibers within the substantia propria. The patients present with relatively rapid onset of a painless, vascularized, nasal or temporal bulbar conjunctival nodule, which can be misinterpreted as a vascularized pinguecula, nevus, squamous carcinoma, or metastasis.
Histopathologic evaluation demonstrates prominent actinic elastosis surrounded by granulomatous response.60
F. Parinaud Oculoglandular syndrome
Parinaud Oculoglandular syndrome is a rare condition, characterized by granulomatous conjunctivitis with regional lymphadenopathy. This syndrome may be caused by a variety of infectious and noninfectious agents, but the most common culprit is Bartonella henselae (cat-scratch disease).61
Cat-scratch disease is the most frequently recognized form of systemic Bartonela henselae infection, which presents with flu-like illness associated with regional lymphadenopathy. Most cases are believed to be transmitted by a scratch or bite from a flea-infested kitten, but direct inoculation with infected flea feces is also possible. Although Parinaud Oculoglandular syndrome is the most common ocular manifestation of cat-scratch disease, other ocular complications include optic neuritis, neuroretinitis, retinochoroiditis, and orbital infiltrates.2,61,62
Serologic testing (indirect immunofluorescence, enzyme-linked immunoassays) is the most cost-effective means for diagnosing cat-scratch disease. A cat-scratch disease antigen skin test can be used also to identify infected patients. Isolation of Bartonella from biopsy specimens is extremely difficult and requires special culture techniques.2,61
Histopathologic evaluation of conjunctival nodules reveals discrete granulomas (which later may become necrotizing) and reactive follicular hyperplasia. The Warthin-Starry silver stain is helpful in demonstrating bacilli within granulomas. Electron microscopy can also demonstrate bacilli in challenging cases (Fig. 22).2,61
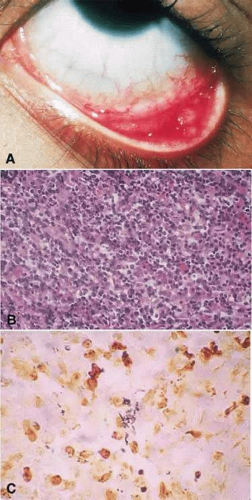 Figure 22. A. Clinical photograph of granulomatous conjunctivitis due to Cat Scratch disease. B. Histopathology shows a dense infiltrate of lymphocytes and epithelioid histiocytes. C. Warthin—Starry silver stain shows numerous pleomorphic bacilli consistent with Bartonella (Rochalimaea) henselae. |
The disease is self limited in immunocompetent patients, one of the reasons why the optimal treatment is debatable. Immunocompromised patients can demonstrate dramatic response to antibiotics, however, such as erythromycin, azithromycin, or doxycycline. Rifampin is often used as adjuvant.61,62
INFECTIOUS
| Bacterial | Mycobacterial | Fungal | Viral |
| Afipia felis | Tuberculosis | Sporotrichosis | EBV |
| Bartonella clarridgeiae | Coccidiomycosis | HSV-1 | |
| Tularemia Syphilis Chlamydia (lymphogranuloma venereum) Yersinia enterocolitica | |||
NONINFECTIOUS
Sarcoidosis
Lymphoma
III. Superior Limbic Keratoconjunctivitis
Superior limbic keratoconjunctivitis (SLK) is an inflammatory disorder that affects the palpebral and bulbar conjunctiva. The condition may arise from mechanical trauma transmitted from the upper eyelid to the superior bulbar and tarsal conjunctiva.69
SLK is usually bilateral, although may be asymmetric, and is more common in women. It exhibits chronic remitting-recurring course, and usually resolves spontaneously. The patients present with a fine papillary reaction on superior tarsal conjunctiva, injection and thickening of the superior bulbar conjunctiva and superior limbus, associated with fine punctate fluorescein and rose Bengal staining. Filamentary keratopathy may be observed on superior cornea.69
Histopathologically, the epithelium is acanthotic with dyskeratosis, loss of goblet cells, and infiltration by neutrophils, lymphocytes, and plasma cells. Epithelial cells may also demonstrate nuclear pyknosis with “snake nuclei” and balloon degeneration of nuclei, and vacuolated cytoplasm.2,69
Treatment includes topical anti-inflammatory agents, bandage contact lenses, punctal occlusion, and thermocauterization or resection of superior bulbar conjunctiva. There is an association between SLK and autoimmune thyroid disease, and the patients should be worked up with appropriate thyroid function tests.2,69,70
Sequelae of Trauma and Inflammation
Cicatrix Formation/ Cicatrizing Conjunctivitis
Trauma and chronic inflammation may result in conjunctival scarring or cicatrix formation. Many of the conditions associated with cicatrizing conjunctivitis and cicatrizing trauma have been described in the preceding section of the chapter.
The unifying histopathologic manifestations of cicatrizing conjunctivitis include epithelial epidermalization with loss of goblet cells, and prominent fibrosis within the substantia propria. Features specific to a certain disease entity may also be observed, such as the presence of immunoglobulin and complement at BMZ in OCP and related disorders, an infiltrate of eosinophils and mast cells in allergic/atopic disease, or a mononuclear infiltrate with occasional granulomatous inflammation in ocular rosacea.
POSTINFECTIOUS CONDITIONS
Trachoma
Adenoviral conjunctivitis
Streptococcal conjunctivitis
Primary herpes simplex conjunctivitis
Corynebacterium diphtheriae conjunctivitis
AUTOIMMUNE AND IMMUNE-MEDIATED HYPERSENSITIVITY DISORDERS
Ocular cicatricial pemphigoid
Sarcoidosis
Medication-induced pemphigoid
Atopic blepharoconjunctivitis
SJS/TEN
Allergic conjunctivitis
Epidermolysis bullosa
Discoid lupus erythematosus
Linear IgA disease
Dermatitis herpetiformis
Graft-versus-host disease
Scleroderma
Paraneoplastic pemphigoid
Lichen planus
Paraneoplastic lichen planus
Wegner’s granulomatosis
PRIOR CONJUNCTIVAL TRAUMA
Radiation
Chemical burns
Surgical trauma
MISCELLANEOUS
Rosacea blepharoconjunctivitis
Porphyria cutanea tarda
Self-induced cicatrizing conjunctivitis
Epithelial Inclusion Cyst
Acquired conjunctival epithelial inclusion cysts usually result in areas of previous surgical or accidental trauma, or as sequelae of chronic conjunctival inflammation with blockage of pseudoglands of Henle. Rarely, these lesions are congenital. Clinical examination reveals a cystic, fluid filled lesion within the superficial conjunctiva (Fig. 23A). Occasionally, white or yellowish concretions are found within the cyst.74,75
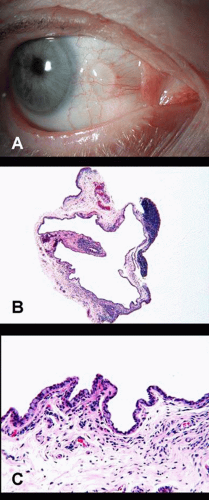 Figure 23. Conjunctival inclusion cyst. A. Cystic lesion filled with clear fluid at the site of prior surgical trauma. (Courtesy Dr. Paul Finger). B. Histopathology discloses a cyst, lined by attenuated conjunctival epithelium with occasional goblet cells. (Stain, H&E) |
Histopathologically, the cyst is lined by nonkeratinized, cuboidal epithelium. When goblet cells are present, mucus accumulates in the cyst. Eosinophilic proteinaceous material (concretions) may occasionally be observed within the lumen (Fig. 23B).
Pyogenic Granuloma
Pyogenic granuloma is a rapidly enlarging, fleshy, red, often pedunculated mass that arises over the site of the injury (surgical trauma, nonsurgical trauma, or inflammation) (Fig 24A).
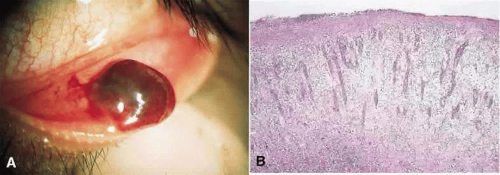 Figure 24. A. Clinical photograph of nodular, fleshy, pedunculated, red mass located in the inferior fornix. B. Histopathology shows radiating pattern of abundant capillaries and fibroblasts amid an edematous stroma. |
The term pyogenic granuloma is a misnomer. It is not granulomatous, nor is it pyogenic. Histopathologically, exuberant granulation tissue is present, characterized by the presence of abundant, radiating, branch-like capillaries, surrounded by fibroblasts and inflammatory cells amidst an edematous stroma (Fig. 24B).76
Topical and intralesional steroids or surgical excision are some of the treatment modalities.
Manifestations of Systemic Disease
Various systemic diseases can have conjunctival manifestations, such as vascular anomalies (ataxia telangiectasia), conjunctivitis and dry eye disease (allergy, atopy, autoimmune disease, hypovitaminosis A), and deposits (metabolic diseases). Some of these conditions have been discussed in the preceding sections of this chapter. In this section, we will address metabolic and nutritional disorders that can affect conjunctiva.
I. Metabolic Diseases
Systemic diseases can affect conjunctiva through abnormal deposition of metabolic substances. Many are lysosomal storage diseases, in which a single enzyme defect results in accumulation of abnormal substances within the lysosomes or lysosome-like intracytoplasmic structures. It is important to note that although conjunctival biopsy is a useful screening test for lysosomal storage diseases, specific diagnosis requires urine testing and a biochemical assays for enzymes or deposits in tears, blood leukocytes, cultured fibroblasts, or amnionic cells. In addition, advances in molecular genetics have made testing for mutations in the specific genes linked to various metabolic diseases possible.
A. Disorders of carbohydrate metabolism: systemic mucopolysaccharidoses
Patients with mucopolysaccharidoses may display varying degrees of facial dysmorphism, skeletal dysplasia, and mental retardation. All are autosomal recessive, except for Hunter syndrome, which is X-linked recessive. Ocular manifestations include progressive corneal clouding, retinal pigmentary degeneration, and optic atrophy.77
Although conjunctival anomalies are not clinically apparent, transmission electron microscopy discloses intracytoplasmic vacuoles filled with fibrillogranular substance within the fibroblasts, endothelial cells, and histiocytes in the superficial substantia propria and within the conjunctival epithelial cells.77 The intracytoplasmic vacuoles contain acid mucopolysaccharide material, which stains with special stains, such as Alcian blue. No extracellular material accumulates.
B. Disorders of lipid metabolism: sphingolipidoses and Batten disease
Sphingolipidoses
The primary lysosomal disorders of complex lipids (gangliosides and sphingomyelin) affect the central nervous system, causing psychomotor retardation. All are autosomal recessive, except for Fabry’s disease, which is X-linked recessive. In addition to psychomotor retardation, Fabry’s disease can also affect the skin, renal, and cardiovascular systems. Ocular manifestations of Fabry’s disease include: corneal clouding, retinal opacification, and optic atrophy. Fabry’s disease manifests in conjunctival and retinal vascular tortuosity, whorl-like corneal opacities (cornea verticillata), periorbital edema, and cortical cataracts. Of note, conjunctival changes in Fabry’s disease may precede corneal deposits and cataracts.77
Conjunctival biopsies show ultrastructural evidence of complex lipid storage, manifesting in concentrically arranged membranous lamellae within Schwann cells, histiocytes, fibroblasts, and endothelial cells. Mucopolysaccharide-like fibrinogranular material may be occasionally demonstrated as well (multiple sulfatase deficiency). In Fabry’s disease, abnormal lipid material accumulates within the adventitial, smooth muscle, and endothelial cells of conjunctival blood vessels.77
Batten Disease (Juvenile Neuronal Ceroid Lipofuscinosis)
The neuronal ceroid lipofuscinoses are a large group of an autosomal recessive, lysosomal storage disorders of childhood, characterized by accumulation of autofluorescent lipopigment granules in most tissues, including brain and retinal neurons. Although the exact pathogenesis and precise characterization of the deposit in Batten disease are unknown, several genes have been identified, some encoding lysosomal enzymes involved in protein metabolism.78,79
The affected patients display progressive psychomotor retardation and visual loss. Ocular findings include “bull’s eye” maculopathy, retinitis pigmentosa fundus, and nonrecordable electroretinogram.78,79
Immunofluorescence studies of the conjunctiva may demonstrate autofluorescent lipopigment granules within the epithelium. Electron microscopy can demonstrate lysosomal inclusions (granular deposits, fingerprint profiles, and curvilinear profiles) within the histiocytes and Schwann cells of peripheral nerves.78,79,80
C. Disorders of mucopolysaccharide and lipid metabolism: mucolipidoses
Mucolipidoses are a group of autosomal recessive metabolic diseases with features common to both mucopolysaccharidoses and sphingolipidoses. Systemic manifestations include mental retardation and facial and skeletal dysmorphism. Ocular manifestations include cornel clouding, due to accumulation of mucopolysaccharides, and retinal grayness with cherry-red spot and optic atrophy, due to accumulation of sphingolipids.
Conjunctival biopsy shows vacuolated fibroblasts, histiocytes, Schwann cells of peripheral nerves, and endothelial cells, filled with both concentrically arranged membranous lamellae, similar to sphingolipidoses, and vacuoles with fibrillogranular material, similar to mucopolysaccharidoses.77
D. Disorders of amino acid metabolism
Cystinosis
Cystinosis is an autosomal recessive disorder manifested by the accumulation of the amino acid cystine within lysosomes of various tissues, due to a defect in transport across the lysosomal membrane.
Three forms have been described:
Infantile (nephropathic) – most common form; dwarfism, progressive renal dysfunction, death at an early age without renal transplantation
Intermediate (adolescent) – less severe renal involvement
Adult – no renal involvement
Eye exam is remarkable for fine, scintillating conjunctival and corneal crystals throughout the corneal stroma and conjunctival substantia propria in all three forms of cystinosis (Fig. 25A). In addition, crystals can occasionally be observed in the trabecular meshwork, iris, ciliary body, choroid, retina, and optic nerve. Photophobia and salt-and-pepper pigmentary retinopathy are also observed in infantile cystinosis.77,81,82
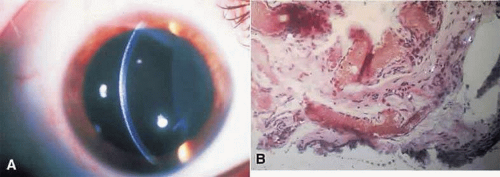 Figure 25. A. Corneal crystals in cystinosis. B. Histopathology shows cystine crystals in substantiapropria stroma. |
Conjunctival biopsy reveals rectangular, square, and hexagonal birefringent crystals, measuring between 0.2 to 2.0 μm in diameter. The crystals accumulate within the epithelial cells and in fibroblasts and histiocytes of subsantia propria (Fig. 25B). The crystals are water-soluble and dissolve during fixation. Therefore, the tissue must be fixed in absolute ethanol and processed rapidly to minimize the aqueous dilution.77,82
Alkaptonuria (Ochronosis)
Alkaptonuria is an autosomal recessive metabolic disorder of tyrosine metabolism, caused by the deficiency of homogentisic acid 1,2-dioxygenase enzyme, which results in an increased level of homogentisic acid in serum. Oxidized homogentisic acid gives a characteristic bluish-black color to urine. Oxidized and polymerized homogentisic acid accumulates within the connective tissues (cartilage, skin, nails, and in the interpalpebral sclera, conjunctiva, and cornea), causing bluish-brown discoloration, referred to as ochronosis.83,84
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


