Chapter 25 Other mesenchymal abnormalities
Rhabdomyosarcoma is discussed in Chapter 24. Every mesenchymal component of the orbit can give rise to sarcomatous tumors, but these are exceedingly rare and will not be discussed. Nevertheless, they are part of the differential diagnosis of rhabdomyosarcoma.
Dysplasias
Fibrous dysplasia of the orbit
Fibrous dysplasia (FD) is rare, characterized by the replacement of normal bone by a cellular fibrous stroma containing islands of immature bone and osteoid which does not remodel along lines of stress into lamellar bone. It has been reported in infancy1 but usually presents in childhood; its onset is insidious and it may remain asymptomatic until adulthood. Progression usually slows in the second or third decade, when “bone maturity” is reached, though growth may continue into the fourth decade in some cases. It is important to distinguish it from meningioma2 and osteosarcoma.
FD may be confined to a single site (monostotic form) or, more rarely, involve multiple bony sites (polyostotic form). Polyostotic FD, which co-exists with cutaneous pigmentation and endocrine abnormalities, is the McCune-Albright syndrome.3 FD is a non-inheritable disorder caused by mis-sense mutations occurring post-zygotically. These result in failure of mesenchymal precursor cells to differentiate normally into osteoblasts resulting in extensive proliferation of fibrous tissue from abnormally differentiated osteoblast precursors. The GSα mutation can be tested for in peripheral blood.4
Clinical features
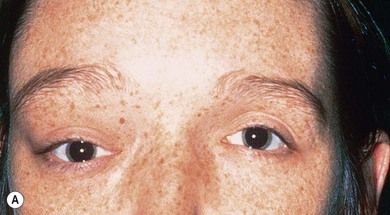
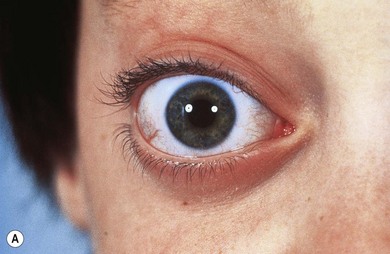
Three-quarters of patients with orbital FD have the monostotic form affecting several contiguous bones but the disease usually remains unilateral. The craniofacial area is affected in 20% of patients, with a predilection for the frontal, sphenoid, and ethmoid bones. Typically, there is a painless, firm, bony swelling with contour distortion; in the orbit, there is an accompanying mass effect. The clinical presentation depends on the predominant wall involved, most commonly the roof, resulting in proptosis and downward displacement of the globe and orbit.5,6 The lacrimal fossa may be affected, mimicking a lacrimal gland tumor.7 Maxillary disease (Fig. 25.1) displaces the eye upwards, with persistent epiphora if the nasolacrimal duct is affected.8 Sphenoid involvement results in optic nerve compression5,6 from narrowing of the optic canal (Fig 25.2) or by an associated sphenoid sinus mucocoele.9 Rarely, involvement of the sella turcica may result in chiasmal compression.10 Other uncommon neuro-ophthalmic complications include cranial nerve palsies,11 trigeminal neuralgia, raised intracranial pressure, and papilledema.11,12 Extensive craniofacial involvement can result in severe cosmetic deformity. Pain may occur, either localized or as a diffuse ipsilateral headache. Visual loss is not uncommon.5,13 Malignant transformation to osteosarcoma, fibrosarcoma, chondrosarcoma, and giant cell sarcoma occurs in 0.5% of cases increasing to 15% with prior radiotherapy.13 The accompanying signs are rapid progression, worsening pain, and infiltration of surrounding structures.
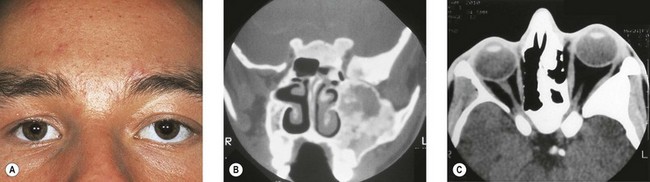
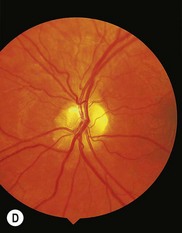
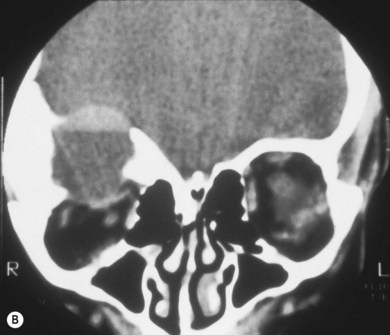
The main radiographic feature of FD is expansion of bone. The lesions may be sclerotic, with a dense ground-glass homogeneity, lytic, with increased lucency, or show a mixed picture with alternating areas of lucency and increased density. Most orbital cases are easily diagnosed since they are sclerotic.5 The main radiologic differential diagnosis includes histiocytosis-X, hyperostotic meningioma, Paget’s disease (both rare in children), and some bone tumors. On magnetic resonance imaging (MRI) there is a correlation between T1 and T2 signal intensity and clinical and pathologic activity of the lesion.14 Occasionally, large cystic lesions form in the orbital wall.5 These may contain blood (Fig. 25.3) and necrotic debris and can be mistaken for aneurysmal bone cysts.13 Involvement of a neighboring sinus may mimic mucocele.13,15 Computed tomography (CT) scanning with the possible addition of MRI is the best modality to evaluate the extent of cranial and orbital involvement. The optic canal and chiasm should be assessed for signs of compression.
Management
When there is little doubt about the diagnosis, initial observation and repeat radiologic assessment is best. If the lesion in the orbital wall is lytic or cystic, biopsy is usually necessary to confirm the diagnosis. Outside the orbit, the risk of malignant change after radiotherapy is high16 and is not used.
Surgery is indicated for cosmetic disfigurement, intractable pain, or optic nerve compression. Since dysplastic bone can be vascular, preoperative blood cross-matching is advisable. Resection of dysplastic bone around the optic canal can reverse visual loss of early compressive optic neuropathy.9,13 Steroids may also be useful.17 When decompressing the optic nerve, rongeurs rather than high speed drills (heat producing) should be used to protect the nerve. Surgery traditionally consisted of de-bulking the lesion. However, the margins of the affected bone are difficult to define clinically and recurrence was common.6 In the last 20 years, there has been a shift toward more radical excision of all diseased bone and immediate facial and orbital reconstruction using bone grafts5,18 by combined ophthalmology/craniofacial teams. Whilst some groups report no visual function deterioration following early optic canal decompression, there have been reports of blindness complicating prophylactic nerve decompression.19 Visual loss is not usually the result of progressive optic canal stenosis but from the rapid expansion of cystic components, FD mucoceles, or hemorrhage.20 Prophylactic optic canal decompression is not indicated and a conservative approach is warranted, reserving optic canal decompression for patients with progressive or sudden deterioration of visual function.21
Bone tumors
Reparative granuloma
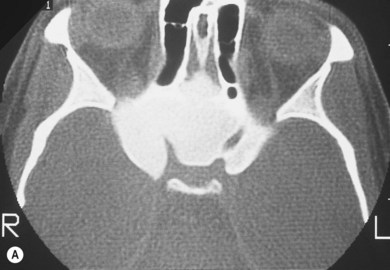
Reparative granulomas (also known as giant cell granulomas) are rare. They affect patients in the first and second decades of life and occur in the mandible, maxilla, and phalanges.13 The lesion may spread to the maxilla, ethmoid,22 and sphenoid bones, involving the orbit (Fig. 25.4) and causing proptosis.23,24 The presentation may be catastrophic if intralesional hemorrhage occurs.13 Histopathologically, there is a spindle cell stroma with profuse hemorrhagic and hemosiderin content. Osteoblastic giant cells are present within the stroma and new bone may be laid down at the edge of the lesion.
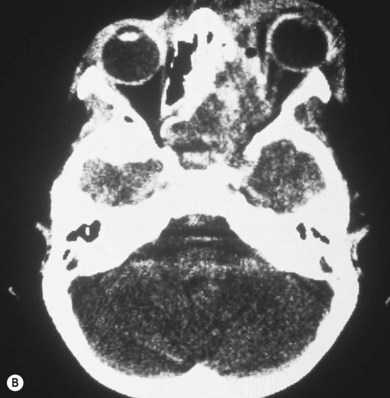
The course is usually benign. Treatment is by surgical curettage; healing occurs by new bone formation. Curettage may need to be repeated or the bony margins resected if the lesion recurs. Radiotherapy is rarely necessary.23
Aneurysmal bone cyst
This uncommon lesion usually affects the metaphysis of long bones or the spine, often preceded by trivial trauma. The skull is affected in less than 1% of cases; about one-quarter of these affect the orbit.25 It is a benign lesion which can usually be differentiated from reparative granuloma by the presence of large blood-filled channels lined by multinucleate giant cells and fibroblasts. However, they can be solid, making this differentiation difficult. The two may coexist.26
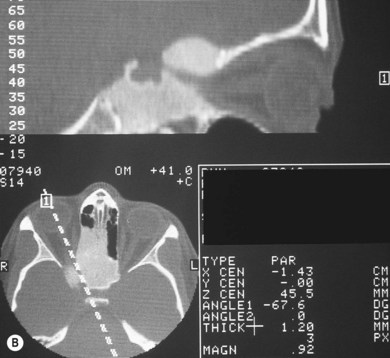
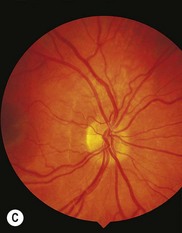
Aneurysmal bone cysts of the orbit (Fig. 25.5) have been periodically reviewed.27,28 Most present in the second decade; there is a 5 : 3 female : male ratio. The history is usually shorter than 3 months with presentations including proptosis, diplopia, ophthalmoplegia,29 ptosis, headache, visual deterioration due to optic nerve compression, nasal congestion,25 epistaxis,30 and epiphora.31 Most cases involve the orbital roof and result in gradually increasing unilateral proptosis and downward displacement of the globe.32 The medial and lateral orbital walls can also be involved. Like reparative granulomas, intralesional hemorrhage may occur, leading to a sudden presentation with signs related to mass effect, occasionally mimicking orbital malignancy in early childhood.33 Large cysts with intracranial extension may give rise to raised intracranial pressure, papilledema,34
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


