14 Orbit
INTRODUCTION
Orbital disease is relatively uncommon but quite diverse in nature. Inflammatory diseases, such as infectious cellulitis, and immunological disorders, such as thyroid ophthalmopathy and idiopathic orbital inflammation or “pseudotumor,” are encountered most often in general clinical practice. However, the soft tissues of the orbit occasionally give rise to a relatively broad spectrum of primary neoplasms, and the orbit can be involved secondarily by systemic lymphoma, metastases from distant primary cancers, or by tumors that arise in neighboring tissues such as conjunctiva, eyelid, paranasal sinuses, or even the intracranial cavity (Fig. 14-1).
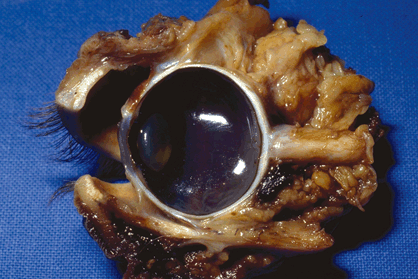
Fig. 14-1. A. Orbital contents. The orbital contents include the eye, optic nerve, smooth and striated muscle, vessels, nerves, fatty and fibrous connective tissue, and the lacrimal gland. They are delimited anteriorly by a fibrous tissue septum and protected by the eyelids.
The essential clinical manifestation of orbital disease is ocular proptosis or exophthalmos (Fig. 14-2A). The eye usually is pushed forward because the orbit is a semi-confined space bounded by bony walls, and most orbital diseases are space-occupying. The proptotic eye may be pushed directly forward (axial proptosis) or in other directions (e.g., down and in) that are determined by, and serve to indicate the location of the orbital lesion. For example, tumors of the lacrimal gland, which is located in the superotemporal orbit, typically push the eye inferomedially as well as forward, while mucoceles of the ethmoid sinus in the medial orbital wall cause lateral displacement. Retraction of the eye or enophthalmos occurs occasionally. Causes of enophthalmos include traumatic blow-out fractures of the orbital floor and sclerosing tumors such as metastatic scirrhous breast carcinoma. Other symptoms and signs of orbital disease include pain, loss of vision, and ocular motility disturbances, which may or may not be conspicuous depending on the underlying cause; some orbital tumors, for example, lymphomas, tend to be well-tolerated. Others, such as metastatic carcinoma, are more likely to be symptomatic.
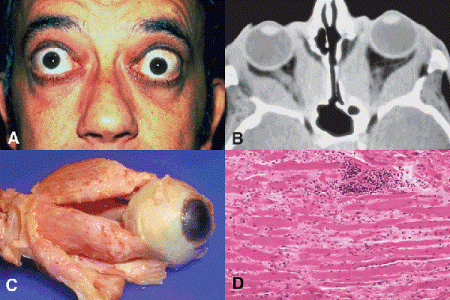
Fig. 14-2. A. Thyroid ophthalmopathy (Graves disease). The patient has bilateral exophthalmos and a characteristic stare. Vertical extraocular muscle imbalance (left hypotropia) reflects fibrosis of left inferior rectus muscle. B. CT shows massive enlargement of extraocular muscles. C. Postmortem exenteration specimen from patient with thyroid optic neuropathy shows massive enlargement of extraocular muscles. D. Extraocular muscle from case seen grossly in Figure C contains patchy foci of chronic inflammatory cells composed largely of lymphocytes. The myofibers are separated by fibrosis. The inflammation in Graves disease characteristically spares the tendons of the extraocular muscles and the orbital fat, features that serve to distinguish the disorder histopathologically from idiopathic orbital inflammatory pseudotumor. (C. Photo by the author. [From Hufnagel TJ, Hickey WF, Cobbs WH, et al. Ophthalmology 1984;91:1411. Courtesy of Ophthalmology.], D. H&E ×50)
ORBITAL INFLAMMATORY DISEASE
Most orbital diseases encountered by ophthalmologists in general practice are inflammatory in nature. The most common inflammatory diseases of the orbit include infectious orbital cellulitides, noninfectious idiopathic orbital inflammation (inflammatory orbital pseudotumor), and Graves disease or thyroid orbitopathy.
Orbital Cellulitis
Orbital cellulitis usually is caused by the extension of a primary sinus infection into adjacent soft tissues of the orbit (Fig. 14-3). Abscesses can form beneath the periosteum (subperiosteal abscess) or in the orbital soft tissues. Patients with orbital cellulitis have signs of acute inflammation including erythema and pain as well as proptosis. Although most orbital cellulitis is caused by bacteria, fungi such as Aspergillus or Mucor occasionally invade the orbit from primary foci in the sinuses. Haemophilus influenzae is an important cause of orbital cellulitis in children but is becoming rare due to immunization. Rarely, a clinical picture resembling orbital cellulitis may be caused by an extensively necrotic intraocular retinoblastoma or melanoma. Orbital involvement has been reported in allergic fungal sinusitis (Fig. 14-4).
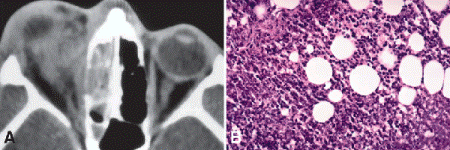
Fig. 14-3. Orbital cellulitis. A. CT scan of patient with orbital cellulitis shows opacification of adjacent infected sinus. Orbital cellulitis often is caused by extension of a primary sinus infection into orbital soft tissues. B. Orbital abscess. An abscess is a focal collection of polymorphonuclear leukocytes. Many of the polys infiltrating the orbital fat are degenerated. Basophilia reflects necrosis. (A. Photo courtesy of Jurij Bilyk, MD, Wills Eye Institute, B. H&E ×100)
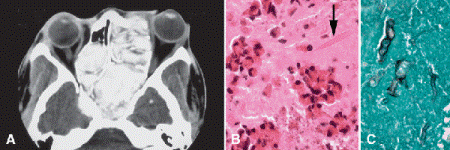
Fig. 14-4. Allergic fungal sinusitis. A. CT scan shows massive involvement and expansion of sinuses by allergic mucin. B. Allergic mucin contains clumps of eosinophils and Charcot-Leyden crystals (arrow). C. Gomori methenamine silver (GMS) stain discloses hyphae of noninvasive fungus within mucin. (B. H&E ×250, C. GMS ×250)
MUCORMYCOSIS
Mucormycosis (zygomycosis) is a potentially lethal opportunistic infection by saprophytic fungi that usually occurs in acidotic persons such as poorly controlled diabetics. The fungus is vasotropic and invades orbital vessels causing thrombosis and necrosis (Fig. 14-5). Histopathology shows both acute and chronic granulomatous inflammation and necrotic tissue. The hyphae are large and nonseptate and are easily identified in standard hematoxylin and eosin-stained sections. Lives occasionally can be saved if an expedient diagnosis is made and antifungal therapy is instituted.
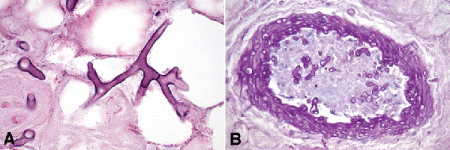
Fig. 14-5. Mucormycosis. A. Necrotic orbital fat contains large branching hyphae. The hyphae are readily seen in routine H&E sections. B. Fungus infiltrates wall and lumen of thrombosed vessel. (A. H&E ×250, B. H&E ×100)
THYROID EYE DISEASE
Thyroid eye disease (thyroid ophthalmopathy, Graves orbitopathy) is the most common cause of unilateral or bilateral exophthalmos (Fig. 14-2). Affected patients have an underlying immunological disorder, which is complex and still poorly understood, that affects both the thyroid gland and orbital structures, especially the extraocular muscles. The exophthalmos is caused by enlargement of the extraocular muscles (Fig. 14-2B,C). The enlarged muscles contain foci of chronic nongranulomatous inflammation and increased quantities of glycosaminoglycans and show endomysial fibrosis (Fig. 14-2D). The tendon of the extraocular muscle and the orbital fat characteristically are noninflamed, a feature that serves to differentiate thyroid ophthalmopathy from idiopathic orbital inflammation (pseudotumor). Visual loss due to compressive optic neuropathy occurs in some patients when the swollen muscle bellies press on the optic nerve in the crowded orbital apex. Corneal complications caused by exposure can also cause visual loss. Graves ophthalmopathy can occur in patients whose thyroid function tests are high, low, or even normal. Most cases are readily diagnosed by the demonstration of enlarged extraocular muscles on computed tomography (CT) or magnetic resonance imaging (MRI) scans. Very few cases are biopsied.
IDIOPATHIC ORBITAL INFLAMMATION
Idiopathic orbital inflammation or pseudotumor is relatively common. Although orbital pseudotumor undoubtedly is an immunological disorder, the details of its immunopathology and the identity of the antigens responsible for inciting the inflammation are still unclear. Idiopathic orbital inflammatory pseudotumor is a diagnosis of exclusion, from both the clinical and histopathologic standpoint. The presence of fungi, bacteria and acid fast organisms, foreign substances, and other specific inflammatory diseases such as Wegener granulomatosis or sarcoidosis must be excluded with special stains or appropriate tests.
Idiopathic orbital inflammation can affect both adults and children who may have either unilateral or bilateral disease. Although occasional cases are relatively asymptomatic and indolent in their course, idiopathic orbital inflammation classically presents explosively with the acute onset of pain, ocular proptosis, muscle paresis, and sometimes visual loss. The inflammation may be localized to single orbital structures such as the extraocular muscles (orbital myositis), the lacrimal gland (chronic dacryoadenitis), or the sclera or episclera (scleritis or episcleritis). A diagnostic trial of systemic corticosteroids may be administered to patients suspected of having idiopathic orbital inflammation, because the disorder is exquisitely sensitive to steroids.
Biopsy is often performed on patients with orbital inflammatory pseudotumor. Microscopy typically discloses a polymorphous infiltrate of inflammatory cells that may contain lymphocytes, plasma cells, eosinophils, macrophages, and occasionally epithelioid histiocytes (Fig. 14-6A,B). Although vessels ringed or cuffed by chronic inflammation can be seen, this reflects diapedesis of lymphocytes, not true vasculitis. Lymphoid follicles and germinal centers occur in some cases, and fibrosis is often extensive. If the lacrimal gland is involved, its acini are destroyed by the inflammatory process and fibrosis replaces the parenchyma. The fibrosis is responsible for the typically eosinophilic appearance of specimens under low magnification light microscopy. This eosinophilia usually distinguishes “pseudotumor” from lymphoid tumors, which are composed of sheets of basophilic cells. Fibrosis is particularly marked in the sclerosing type of pseudotumor, which may be a subset of IgG4-related sclerosing disorders. Follicular lymphoid hyperplasia is considered to be an orbital lymphoid tumor by ophthalmic pathologists; it should not be called an inflammatory pseudotumor.
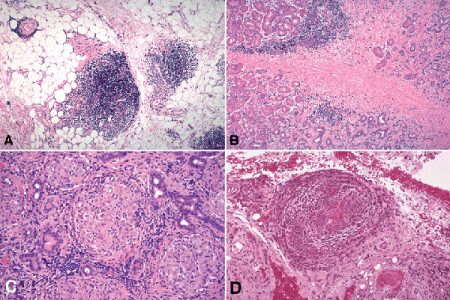
Fig. 14-6. A. Idiopathic orbital inflammation, orbital fat (idiopathic inflammatory pseudotumor). The orbital fat contains patchy foci of chronic inflammatory cells. The polymorphous inflammatory infiltrate was composed largely of lymphocytes and plasma cells. B. Idiopathic orbital inflammation with fibrosis, lacrimal gland (sclerosing idiopathic inflammatory pseudotumor). A few residual ductules persist in the eosinophilic, chronically inflamed scar tissue that has replaced the lacrimal gland. Patchy foci of chronic inflammatory cells are present. Higher magnification disclosed a polymorphous inflammatory infiltrate composed largely of lymphocytes and plasma cells. C. Sarcoidosis. Atrophic parenchyma of lacrimal glands contains multiple discreate noncaseating granuloma consistent with sarcoidosis. D. Wegener granulomatosis. Granulomatous vasculitis involves orbital vessel. Foci of necrosis were present. (A. H&E ×25, B. H&E ×25, C. H&E ×50, D. H&E ×50)
Lacrimal gland biopsy typically discloses discrete noncaseating granulomas composed of epithelioid histiocytes and giant cells in sarcoidosis, which is another diagnosis of exclusion (Fig. 14-6C). Lacrimal gland biopsy occasionally is performed to confirm the diagnosis of sarcoidosis histopathologically.
If true vasculitis, granulomatous inflammation, and focal necrosis are observed, the patient may have Wegener granulomatosis. About one third of patients with Wegener granulomatosis have ophthalmic manifestations during the course of their disease, and they may present with ocular findings such as orbital infiltration or peripheral corneal ulceration. Clinical suspicion is important because the classic histopathologic features of Wegener granulomatosis are found in few orbital biopsies (Fig. 14-6D). Most patients have concurrent sinus disease. The cytoplasmic antineutrophilic cytoplasmic antibody (c-ANCA) test is a helpful diagnostic adjunct but may be negative in the early stages of the disease.
ORBITAL TUMORS
A wide variety of benign and malignant primary neoplasms are spawned by the tissues of the orbit. Most of these tumors are relatively rare. Secondary involvement of the orbit by blood-borne metastases, systemic lymphoproliferative disorders, or direct invasion from contiguous structures also occurs. Children and adults are affected by different spectra of orbital tumors.
Orbital Tumors in Adults
Primary orbital tumors that are encountered fairly often in adults include cavernous hemangioma, schwannoma, fibrous histiocytoma (FH) or solitary fibrous tumor (SFT), hemangiopericytoma, epithelial tumors of the lacrimal gland, and lymphoid tumors. Cavernous hemangioma and lymphoid tumors are encountered most often.
LYMPHOID TUMORS
Orbital lymphoid tumors comprise a spectrum that includes polyclonal reactive lymphoid hyperplasias and malignant lymphomas composed of clonal proliferations of lymphoid cells (Figs. 14-7 and 14-8). Immunophenotypic analysis has shown that most lesions previously classified light microscopically as atypical lymphoid hyperplasias are low-grade lymphomas. Lymphoma is also discussed in Chapter 5.
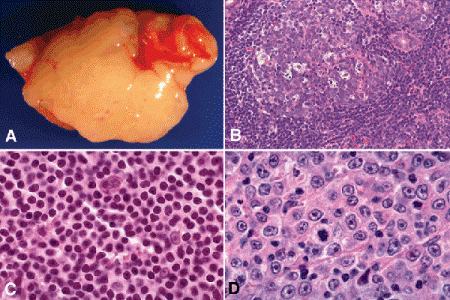
Fig. 14-7. Lymphoid tumors. A. Cut surface of soft orbital lymphoma has uniform salmon-yellow-color. B. Follicular lymphoid hyperplasia. Small well-differentiated lymphocytes surround a germinal center. The lighter cells in the germinal center are tingible body macrophages, which are antigen presenting cells. Mitoses normally are found in germinal centers. The germinal center and adjacent mantle zone are composed largely of B lymphocytes. C. Diffuse non-Hodgkin lymphoma, well-differentiated lymphocytic type. Infiltrate is composed of monotonous monomorphic sheet of small well-differentiated lymphocytes. Immunophenotypic studies disclosed a monoclonal population of B lymphocytes. D. Diffuse large cell lymphoma. This obviously malignant tumor is composed of large, atypical immunoblastic B cells that have prominent nucleoli. Apoptotic cells and mitoses are present. (B. H&E ×100, C. H&E ×250, D. H&E ×250)
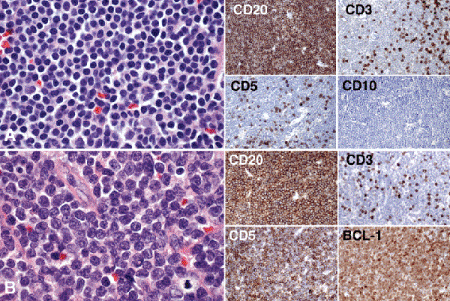
Fig. 14-8. A. Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma). Lymphoma is composed of diffuse infiltrate of small, well-differentiated B lymphocytes. IHC panel at right shows that neoplastic B cells express B cell marker CD20 but are negative for CD3, CD5, and CD10. More than half of ocular adnexal lymphomas are MALT lymphomas. B. Mantle cell lymphoma. The neoplastic B cells comprising mantle cell lymphoma usually have irregular cleaved nuclei. The CD20-positive B cells coexpress T cell marker CD5 but are negative for CD3. Positive immunoreactivity for BCL-1 (cyclin D1) confirms the diagnosis. Mantle cell lymphoma has a poor prognosis. (A. main figure, H&E ×250; insets, IHC for CD20, CD3, CD5, and D10, all ×100; B. main figure, H&E ×250; insets, IHC for CD20, CD3, CD5 and bcl-1, all ×100)
Orbital lymphoma usually is a disorder of older patients that is diagnosed on average at age 60 years. The onset of orbital lymphoma is usually insidious. Orbital lymphoid tumors typically present with painless, well-tolerated proptosis, because the tumor has no fibrous stroma, is soft and pliable, and molds to the globe and other orbital structures. Some patients may present with a conjunctival mass or patch of salmon-colored tissue on the epibulbar surface or fornix (Fig. 5-29). The tumor’s characteristic salmon hue reflects the presence of many fine capillaries (Fig. 14-7A). The lymphoid infiltrate is often delimited sharply by tissue planes. The latter are evident on imaging studies as linear margins. Lymphomas diffusely infiltrate and thicken the lacrimal gland, which drapes around the globe assuming an appearance that Jakobiec has facetiously termed a “pregnant pancake.” The latter contrasts with epithelial neoplasms of the lacrimal gland, which typically are rounded. Ninety percent of orbital lymphomas involve the superior part of the orbit behind the orbital septum. More than 40% involve the lacrimal gland and often affect its palpebral lobe. Bone destruction is very rare except in multiple myeloma. When extraocular muscles are involved, usually a single muscle is affected. Motility remains normal because the lymphoma does not stimulate fibrosis.
Ocular lymphomas are extranodal by definition because the orbit lacks lymphatics and lymph nodes, and most are diffuse, as well. About two thirds of ocular adnexal lymphoid tumors are malignant non-Hodgkin lymphomas (NHLs) composed of a monoclonal proliferation of B lymphocytes, and most of the these are low grade lesions composed of well-differentiated lymphocytes. In recent years, pathologists have used the WHO classification to classify lymphomas. Types of NHL in the WHO classification that typically affect the ocular adnexa include extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (often called MALT lymphoma), follicular lymphoma, mantle cell lymphoma, diffuse large cell lymphoma, and small lymphocytic lymphoma.
More than half of ocular adnexal lymphomas are MALT lymphomas (Fig. 14-8A). About 23% are follicular lymphomas, 5% mantle cell, and 4% small lymphocytic lymphoma (SLL/CLL). These four entities are very difficult to differentiate using routine light microscopy alone, but distinguishing them is important clinically because they differ greatly in prognosis and the proper choice of therapy requires accurate classification. The cells of extranodal marginal zone or MALT lymphoma are CD20 positive but are negative for CD3, CD5, and CD10 (Fig. 14-8A). Mantle cell lymphoma (Fig. 14-8B) is usually widely disseminated on presentation and has a poor prognosis. Its CD20-positive B cells co- express T cell marker CD5 and bcl-1 (also called cyclin D1). The cells comprising the neoplastic follicles of follicular lymphoma stain with CD20, CD10, and bcl-2. About one third of patients who have orbital lymphomas have, have a history of, or will develop systemic lymphoma. All patients with ocular adnexal lymphoid tumors need to be evaluated by a hematologist-oncologist. Some patients with polyclonal lymphoid proliferations are a risk for systemic lymphoma.
Lymphoid tumors of the orbit and ocular adnexa are treated with external beam radiotherapy with appropriate eye shielding if there is no evidence of systemic disease. Chemotherapy or immunotherapy should be used if extraocular systemic lymphoma is present. This may be supplemented with adjuvant radiotherapy if ocular regression is subtotal.
VASCULAR LESIONS
Cavernous Hemangioma
Cavernous hemangioma is probably the most common primary orbital neoplasm. This benign vascular tumor typically occurs in middle-aged women. Cavernous hemangiomas cause low-grade proptosis and usually are well-tolerated, sparing vision and ocular motility. Some cases are discovered incidentally when imaging is performed for headache or other indications (Fig. 14-9A). These benign vascular tumors are well-circumscribed, encapsulated lesions with a pebbly surface that appear dusky-red or purplish-blue grossly (Fig. 14-9B). They are composed of large round or oval vascular spaces lined by endothelium, which are separated by thick septa of fibrous tissue that may contain smooth muscle (Fig. 14-9C,D). Cavernous hemangiomas show little contrast enhancement on imaging studies because their circulation is relatively stagnant. This feature helps to distinguish them from the much rarer encapsulated vascular tumor hemangiopericytoma, which enhances vividly.
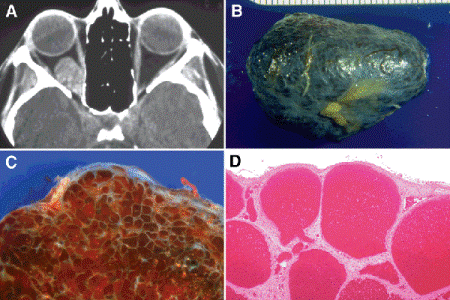
Fig. 14-9. Cavernous hemangioma, orbit. A. CT scan shows characteristically well-circumscribed oval mass within muscle cone. The differential diagnosis of a well-circumscribed orbital tumor also includes schwannoma, hemangiopericytoma and SFT. B. After fixation, the well-circumscribed, encapsulated tumor appears bluish-purple in color and has a pebbly surface. C. The benign encapsulated tumor is composed of large blood-filled vascular channels separated by fibrous septa that are evident grossly. D. The large vascular channels comprising the benign vascular tumor are lined by a single layer of endothelial cells. The fibrous septa separating the vessels often contain smooth muscle. (D. H&E ×25)
Hemangiopericytoma
Hemangiopericytomas is a relatively rare orbital lesion. Hemangiopericytoma was originally thought to be a soft tissue tumor that arose from pericytes in the walls of capillaries. Large branching sinusoidal vessels with a “stag-horn” configuration are a classic histopathologic feature, and the reticulin stain shows that its constituent cells are totally enveloped by basement membrane (Fig. 14-10). About 15% of patients who were diagnosed as having orbital hemangiopericytoma at the Armed Forces Institute of Pathology developed distant metastases. The histologic appearance of the tumor did not always predict metastatic potential, as metastatic disease occasionally developed in patients with benign-appearing tumors. In recent years, authorities have stressed that many other neoplasms may show a branching staghorn hemangiopericytomatous vascular pattern. It is now believed that many tumors diagnosed as hemangiopericytomas in the past probably would now be classified as solitary fibrous tumors (SFTs) (Fig. 14-13).
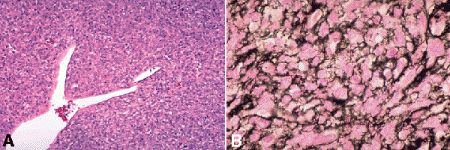
Fig. 14-10. Hemangiopericytoma. A. Highly cellular tumor contains branching staghorn sinusoidal vessel. B. Reticulin stain highlights basement membrane encompassing neoplastic pericytes. (A. H&E ×50, B. Wilder reticulin ×250)
Lymphangioma
Lymphangiomas and capillary hemangiomas (hemangioma of infancy) and are the most common vascular tumors of the orbit in children (Fig. 14-11). Lymphangiomas are poorly circumscribed choristomatous lesions composed of large endothelial-lined channels filled with lymph or serosanguineous fluid (Fig. 14-11A,B). Focal lymphoid infiltrates in the contiguous stroma, which may contain germinal centers, serve to differentiate lymphangioma from cavernous hemangioma, particularly in cases where there has been secondary hemorrhage into the lymphatic vessels. Acute enlargement of lymphangioma is often caused by intralesional hemorrhage, which can produce a blood-filled “chocolate cyst” (Fig. 14-11A). Lymphangiomas also may enlarge during upper respiratory infections, presumably because their lymphoid component undergoes hyperplasia. Lymphangiomas are usually quite infiltrative in their growth pattern, and they do not undergo spontaneous involution. Surgical removal may be quite difficult. Orbital lymphangiomas stain positively with the D2-40 immunohistochemical (IHC) stain for lymphatic endothelial cells.
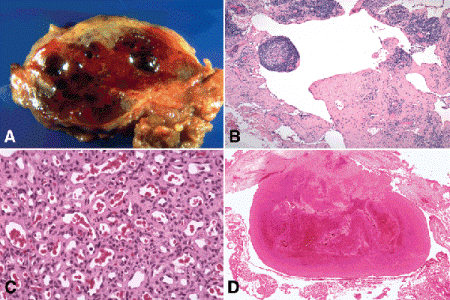
Fig. 14-11. Vascular lesions. A. Lymphangioma with secondary hemorrhage. Blood-filled chocolate cysts caused by secondary hemorrhage into lymphangioma are evident in this gross specimen. Many lymphangiomas contain blood. Lymphangiomas typically are unencapsulated and show an infiltrative growth pattern. B. Lymphangioma, orbit. Lymphoid channels comprising lymphangioma vary markedly in size and shape. They appear empty or contain serous fluid if intralesional hemorrhage has not occurred. Lymphoid foci in intervascular septa help to differentiate a lymphangioma with secondary hemorrhage from a cavernous hemangioma. Lymphangiomas typically are unencapsulated and have an infiltrative growth pattern. C. Capillary hemangioma. Mature capillary hemangioma is composed of a plexus of capillary caliber vessels. Early lesions often are composed predominantly of a solid sheet of endothelial cells. Vascular lumina develop and become progressively ectatic at the lesion matures. D. Thrombosed varix, orbit. A varix is a dilated or ectatic vein. An organized thrombus adheres to the wall of this orbital varix. (B. H&E ×10, C. H&E ×100, D. H&E ×5)
Capillary (Infantile) Hemangioma
Involvement of eyelid skin by a capillary hemangioma is readily apparent as a bright red strawberry nevus. Infantile hemangiomas confined to the orbit are more subtle and may present with proptosis and bluish discoloration of the skin. Poorly circumscribed and unencapsulated, these benign vascular tumors are composed of lobules of capillary-caliber vessels or sheets of capillary endothelial cells (Fig. 14-11C). Hemangiomas in very young infants tend to be quite cellular; capillary lumens appear and enlarge as the lesions mature. Although infantile hemangiomas eventually involute spontaneously, they may be a major cosmetic blemish and often produce potentially reversible visual loss (amblyopia) by causing corneal astigmatism or by occluding the pupil. Nonsurgical therapy includes intralesional injection of steroids, interferon α-2a and systemic propranolol.
Other vascular lesions that affect the orbit include varices and arteriovenous malformations. Patients with orbital varices often show variable proptosis that depends on head position and is increased by a Valsalva maneuver. Histopathology shows a markedly dilated vein that may contain a thrombus (Fig. 14-11D). Intravascular papillary endothelial hyperplasia may develop in the thrombosed varix.
Neurogenic Tumors
Neurogenic tumors of the orbit include schwannomas, neurofibromas, amputation neuromas, and malignant peripheral nerve sheath tumors.
Schwannoma
Schwannoma or neurilemmoma is a well-circumscribed, encapsulated orbital tumor of adults composed of a neoplastic proliferation of Schwann cells, the perineural cells that form the myelin sheaths around axons in peripheral nerves (Fig. 14-12A). Many orbital schwannomas arise from sensory branches of the ophthalmic division of the trigeminal nerve, and may be painful. Histopathology shows an encapsulated spindle cell neoplasm composed of cells with eosinophilic cytoplasm that have indistinct cellular borders and bland elongated oval nuclei. Two growth patterns are recognized. Tumors with the solid Antoni A pattern show bands of nuclear palisading and structures called Verocay bodies (Fig. 14-12B). The Antoni B pattern is marked by a loose myxomatous background. Schwannomas are immunoreactive with neural markers such as S-100 protein and CD57.
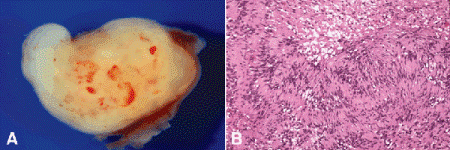
Fig. 14-12. Schwannoma (neurilemoma). A. Cut surface of well-circumscribed schwannoma is yellow-tan in color. B. Antoni A portion of benign peripheral nerve sheath tumor (below) is composed of highly regimented fascicles of bland spindle cells that show nuclear palisading and enclose tangles of fibrillary processes called Verocay bodies. Small focus of looser myxoid tumor (Antoni B pattern) is seen above. Schwannomas are well-circumscribed, encapsulated tumors that are associated with a peripheral nerve. (B. H&E ×100)
Neurofibroma
Isolated neurofibromas cause diffuse enlargement of the affected nerve and contain axons. They are pseudoencapsulated. Schwannomas and neurofibromas are often grouped together under the designation peripheral nerve sheath tumor. Schwannomas and isolated neurofibromas are more prevalent in patients who have neurofibromatosis. Plexiform neurofibromas and diffuse neurofibromas are characterstic manifestations of NF-1. Malignant peripheral nerve sheath tumors are encountered rarely.
Mesenchymal Tumors
A variety of rare mesenchymal tumors of fibrous, fibro-osseous, smooth muscle, adipose tissue, or cartilaginous derivation occur in the orbit. Other extremely rare primary orbital tumors include endodermal sinus tumor, alveolar soft part sarcoma, granular cell tumor, paraganglioma, primary orbital carcinoid, primary orbital melanoma, retinal anlage tumor, neuroepithelioma, ectomesenchymal tumor, malignant rhabdoid tumor, and primitive neuroectodermal tumor.
Fibrous Histiocytoma and Solitary Fibrous Tumor
FH is said to be the most common mesenchymal tumor of the orbit in adults. Typically well-circumscribed but non-encapsulated, FH is composed of spindle cells arranged in a characteristic whirling, pinwheel or storiform pattern. Benign, locally aggressive and rare malignant variants of FH are recognized. Although benign and locally aggressive FH cannot metastasize, total excision is recommended to prevent recurrence. Many tumors that were diagnosed as FH in the past probably would be classified today as solitary fibrous tumors (SFTs), a newly recognized orbital spindle cell neoplasm that shares many features with FH (Fig. 14-13). The cells in SFT are arranged in a “patternless pattern” and are immunoreactive for CD-34, CD99, and bcl-2.
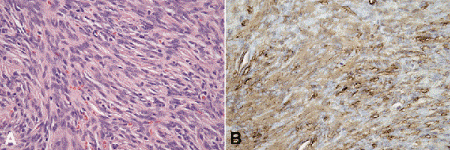
Fig. 14-13. Solitary fibrous tumor, orbit. A. Spindle cells comprising SFT are arranged in a “paternless pattern.” B. Tumor cells are immunoreactive for CD34 (shown here), CD99 and bcl-2. (A. H&E ×100, B. IHC for CD34 ×100)
Fibro-osseous Lesions
Common fibro-osseous lesions of the orbital bones include fibrous dysplasia, juvenile psammomatoid ossifying fibroma, and ivory osteoma. Ivory osteoma is the most common bony lesion in adults. Fibrous dysplasia generally occurs in the first two decades and may spread across suture lines to involve multiple orbital bones. The affected bones have a “ground glass” appearance in CT bone windows (Fig. 14-14A,B,C–D). Fibrous dysplasia is composed of irregular trabeculae of immature woven bone surrounded by fibrous stroma. The bony trabeculae are not rimmed by osteoblasts and are often shaped like Chinese characters.
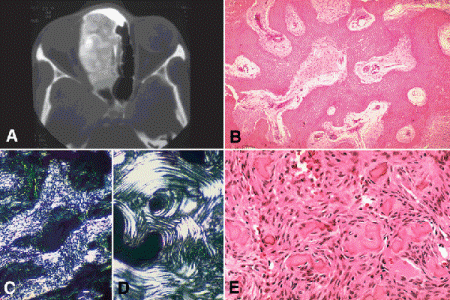
Fig. 14-14. Fibro-osseous lesions. A. Fibrous dysplasia. Lesion of nasal bones has “ground glass” appearance on CT scan. Fibrous dysplasia may spread across suture lines to involve multiple orbital bones. B. Fibrous dysplasia. Fibrous stroma contains irregular trabeculae of immature woven bone, which are not rimmed by osteoclasts. Fibrous dysplasia represents an arrest in the maturation of bone. C. Polarization microscopy of fibrous dysplasia discloses an irregular interweaving pattern of collagenous matrix that resembles the fibers in woven cloth. D. In contrast, collagen fibers disclosed by polarization in mature lamellar bone form highly regular, parallel lamellae. E. Psammomatoid ossifying fibroma. The cellular stroma contains spindle cells and small spicules of bone called ossicles that can be confused with the psammoma bodies of meningioma. This benign lesion found in young individuals has an expansile growth pattern and behaves more aggressively than fibrous dysplasia. It usually does not cross suture lines and is restricted to a single orbital bone. (B. H&E ×25, C. H&E with crossed polarizers ×50, D. H&E with crossed polarizers ×50, E. H&E ×100)
Juvenile ossifying fibroma is a more aggressive expansile lesion that usually is restricted to a single bone. Radiographically, it has a sclerotic margin and a less radiodense center. The cellular fibrous stroma of the psammomatoid variant of juvenile ossifying fibroma contains bony spicules that can be confused with psammoma bodies and lead to the misdiagnosis of meningioma (Fig. 14-14E).
The orbital bones occasionally are affected by a perplexing group of rare osseous lesions that contain giant cells including aneurysmal bone cyst, giant cell reparative granuloma, giant cell tumor, the brown tumor of hyperparathyroidism, and eosinophilic granuloma (see below). Osteogenic sarcoma and other soft tissue sarcomas can arise after radiotherapy for retinoblastoma.
Lacrimal Gland Lesions
The lacrimal gland is a minor salivary gland that is located in a bony fossa behind the superotemporal orbital rim (Fig. 14-15). Lacrimal gland tumors constitute only 10% to 15% of orbital lesions. Most lacrimal gland lesions encountered in nonreferral clinical practice are inflammatory or lymphoid tumors, which are at least five times more prevalent than primary epithelial tumors. Epithelial neoplasms of the lacrimal gland are quite rare, but they are important because about half are highly malignant tumors that are potentially lethal.
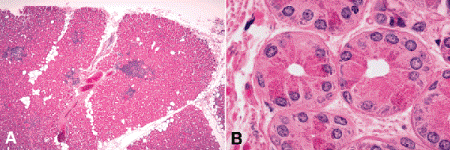
Fig. 14-15. Lacrimal gland. A. Fibrofatty stroma separates multiple lobules composed of glandular acini. Patchy foci of chronic inflammation are present. B. Lacrimal gland, acini. Inner layer of tall columnar secretory cells and a relatively inconspicuous discontinuous outer layer of contractile myoepithelial cells comprise acini of lacrimal gland. Large intensely eosinophilic secretory granules called zymogen granules are found in the apical cytoplasm of the secretory cells. (A. H&E ×10, B. H&E ×250)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


