Optic Pathway Gliomas
Adam S. Hassan
Victor Elner
First described by Scarpa in 1816,1 optic pathway gliomas are astrocytic neoplasms of the optic nerve, chiasm, tracts, and radiations. There are two distinct types of optic pathway glioma. The classical type, which occurs chiefly in childhood, is frequently associated with neurofibromatosis type-1 (NF-1) and is characterized by relatively slow growth, which may or may not be progressive. The second, much rarer type of optic pathway glioma occurs mainly in adults. This malignant neoplasm exhibits rapid growth and is almost always lethal. This chapter will concentrate on the classical type and briefly review what is known of the malignant variant.
Background
Gliomas of the optic nerve and intracranial optic pathways comprise only 1.5% to 3.5% of orbital tumors2,3 and 1% of all intracranial neoplasms,4,5 respectively. In children, however, optic gliomas account for approximately 17% of all orbital tumors6 and 3% to 5% of all primary intracranial neoplasms.7,8 Representing more than 50% of intrinsic tumors of the optic nerve,9 gliomas affect only the intraorbital optic nerve in 15% to 25% of cases, whereas 60% involve the chiasm and 25% involve other intracranial structures.10,11
Approximately 90% of optic gliomas present in the first two decades of life with more than 75% occurring within the first decade.12,13,14,15,16 The median age of patients with anterior visual pathway gliomas is 6.5 years,17 but these tumors may present in adulthood and behave in a similar fashion.18 Optic nerve gliomas are almost always sporadic and unilateral except in the setting of NF-1 in which they may be bilateral and familial.6,19,20 When presenting early in life, the gliomas tend to be larger and more frequently exhibit chiasmatic and diencephalic involvement.10,21 In addition to NF-1, systemic associations with cases of optic gliomas have been reported with colonic polyposis22 and tuberous sclerosis.23
Manifestations of optic nerve glioma
Clinical Findings
Most patients with optic gliomas exhibit symptoms or signs of visual loss. In patients noting visual impairment, the loss is described as painless and gradual. However, rapid loss of vision may occur due to bleeding within the tumor.27 Approximately 75% of the patients with measurable visual loss present with acuity of <20/40.10 A relative afferent pupillary defect is typically present and color vision may be abnormal.
Painless proptosis is common and frequently the presenting sign in patients with orbital gliomas10,28 (Figs. 1 and 2). Proptosis may also precede visual loss.21 The proptosis is generally mild to moderate, nonpulsatile, and axial. Ocular motility may be altered by sensory-induced strabismus24,29 due to unilateral visual loss. Bulky intraorbital tumors may cause mechanical restriction28 of eye movement while increased intracranial pressure due to the growth of posteriorly located tumors can cause a paralytic strabismus.
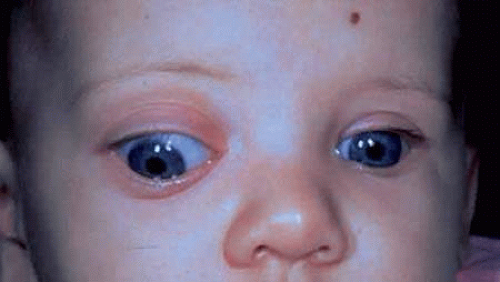 FIGURE 1. Five-month-old infant presenting with progressive proptosis caused by an optic pathway glioma. There is mechanical restriction of the motility of the affected right eye. |
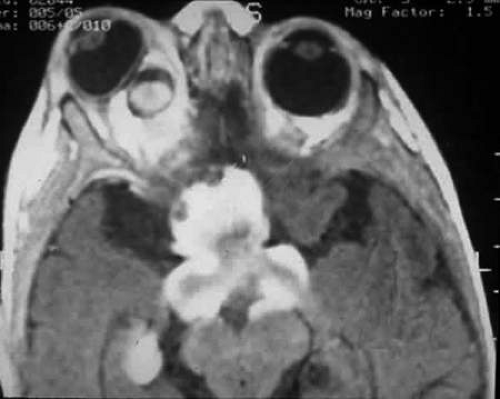 FIGURE 2. Contrast-enhanced T1-weighted axial MRI of the orbits of the patient in Figure 1, which demonstrates a large optic pathway glioma with posterior extension into the optic tracts and radiations. (Courtesy of Orlando Ortiz, MD, and Jeffrey Hogg, MD). |
Optic atrophy, of variable degree, is present in greater than half of the cases of intraorbital or chiasmal gliomas, which also may cause optic disc edema.10 Retinal vascular features resulting from glioma compression of the central retinal vein include venous stasis retinopathy, retinal venous occlusion, and neovascular glaucoma.30,31,32 Optociliary shunt vessels also have been reported, albeit less commonly than in cases of optic nerve sheath meningioma.30,31 Ocular compression may reduce axial length and induce refractive errors and retinal striae.28
A-scan ultrasonography on optic nerve gliomas reveal homogeneous acoustic density with low-to-medium internal reflectivity, which contrasts with the findings seen in optic nerve sheath meningiomas, wherein irregular acoustic density is associated with medium to high reflectivity. The B-scan sonogram characteristically exhibits a fusiform mass altering the optic nerve shadow.33 In the minority of cases of optic nerve glioma, increased subarachnoid fluid within the nerve sheath may be found with the 30-degree test performed using the A-scan mode. Although the test is usually negative because of solid thickening of the nerve by tumor, it may be positive in patients with increased intracranial pressure, because excess subarachnoid fluid is redistributed as the patient shifts gaze from primary fixation to 30° from fixation.34
Visual field alterations are variable and depend on the location along the visual pathway, as well as the extent of cross sectional involvement. Thus, diverse patterns of visual field deficits include central, cecocentral, altitudinal, and bitemporal scotomas, as well as nonspecific constriction. In patients with normal acuity, visual evoked potentials (VEP) demonstrate 100% sensitivity and 60% specificity in identifying and 83% sensitivity in assessing damage due to optic pathway gliomas.35,36
Radiologic Findings
Computed tomography (CT) and magnetic resonance imaging (MRI) are the mainstay for diagnostic imaging of optic pathway gliomas. CT or MRI reveals optic nerve thickening or fusiform nerve enlargement resulting in a kinked, S-shaped profile due to tumor proliferation confined to the nerve sheath30,31 (Fig. 3). Cystic spaces due to fluid imbibed by mucinous secretions of the tumor cells may be evident on imaging, particularly in patients with rapidly progressive proptosis.37,38 Common in meningiomas, calcification is rarely seen in CT scans of optic gliomas.21 Posterior extension of optic nerve gliomas may cause enlargement of the optic foramen that is best visualized by CT scanning, but may also be observed on plain radiographs. Generally, slow growth of gliomas causes remodeling of the optic foramen with a resulting enlargement of the foramen while preserving a corticated margin.28 (Fig. 4) Intracranial extension of the tumor is not always present when the optic canal is enlarged, because reactive arachnoid and dural hyperplasia may cause this change.39,40
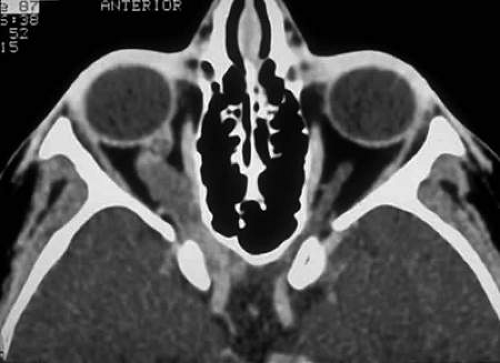 FIGURE 3. Contrast-enhanced axial CT scan showing an enlarged, kinked optic nerve with posterior extension of the glioma through the optic canal. Note the widening of the optic canal on the right. (Courtesy of Orlando Ortiz, MD, and Jeffrey Hogg, MD). |
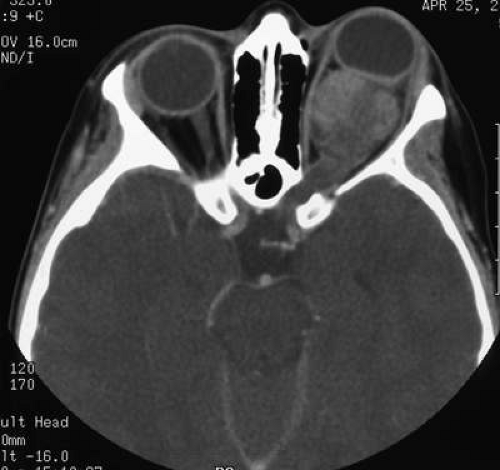 FIGURE 4. Optic nerve glioma unassociated with NF-1, which demonstrates enlargement of optic foramen. Bone of optic canal shows remodeling due to slow growth of tumor. |
Initially complementing CT scanning in the evaluation of optic pathway gliomas, MRI scanning has replaced CT scanning as a better method to delineate the extent of tumor involvement and tumor progression. Because patients with optic pathway gliomas are frequently monitored by sequential imaging studies, MRI has the benefit of also limiting total X-ray dosage. (Fig. 5 A and B). T1 relaxation times are normal to slightly prolonged, whereas T2 relaxation times are prolonged. The T1-weighted images of the tumor are slightly hypointense when compared to normal brain tissue. T2 images of the tumors contrast with brain tissue due to their prolonged relaxation times, thereby permitting assessment of posterior tumor extension.41 Sheath thickening due to reactive arachnoid hyperplasia does not enhance as well as the neoplastic proliferation of meningothelial cells found in the optic nerve sheaths of meningiomas.42 To improve tumor delineation, gadolinium enhancement combined with fat suppression may be used.
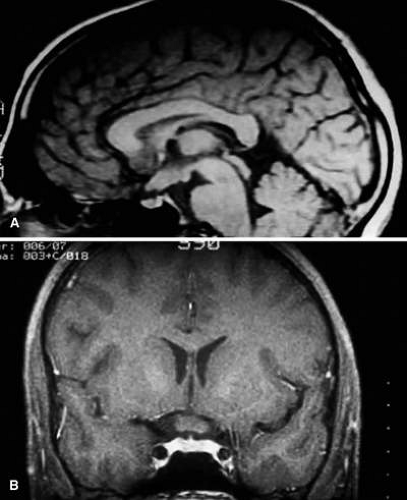 FIGURE 5. A. T1-weighted sagittal MRI of a patient with NF-1, which demonstrates enlarged optic chiasm consistent with optic glioma. B. T1-weighted coronal image with gadolinium and fat suppression in the same patient, which demonstrates the chiasmal glioma. |
Manifestations of intracranial optic nerve glioma
Clinical Findings
More than 50% of optic gliomas exhibit chiasmal involvement, with approximately half of these extending into adjacent structures, most commonly the hypothalamus or third ventricle.10 When gliomas are intracranial, symptoms may be referable to the effects of increased intracranial pressure or direct hypothalamic involvement. They include headache, vomiting, somnolence, or thirst due to diabetes insipidus. Examination may also reveal findings of precocious puberty.45
Due to tumor involvement of the chiasm and other portions of the intracranial optic pathway, patients with these tumors often present with bilateral visual dysfunction.17,24,25,29,46 However, proptosis may be subtle or absent. Vertical, horizontal, rotary, and see-saw nystagmus may occur and may even be a presenting sign before visual loss occurs. Thus, children with spasmus nutans or unusual patterns of nystagmus require MRI study and follow-up imaging to exclude the diagnosis of chiasmal glioma.47,48,49,50,51
Optic disc swelling may result from direct glioma extension into the optic nerves, whereas papilledema, more frequently bilateral, may be induced by the mass effect of intracranial tumor growth. Papilledema may also result from ventricular obstruction.
Visual field defects may be irregular and vary depending on the site and extent of glioma involvement of the visual pathways, as well as the degree of dysfunction induced by the tumor.52,53,54,55,56 The absence of a visual field defect should not be interpreted as an absence of tumor in the visual pathway because nerve axons may be unaffected as they pass through areas of glioma involvement.28 This indicates the importance of neuroimaging in these cases.
Patient age influences the constellation of symptoms and signs when intracranial involvement is present. According to Wisoff,57 there are three age-dependent characteristic clinical patterns in patients harboring large chiasmal-hypothalamic tumors:
<2 years: visual dysfunction, macrocephaly, and failure to thrive
2 to 5 years: endocrine dysfunction most common presenting sign
>5 years and young adults: visual complaints as the primary feature
Hypothalamic involvement may cause endocrinologic abnormalities such as, diabetes insipidus, panhypopituitarism, girdle-type obesity, gigantism, and dwarfism.58,59 With associated pituitary involvement, precocious puberty and growth hormone deficiency may be present.60,61 Extensive growth may cause other neurologic manifestations of epilepsy, hemiparesis, and the diencephalic syndrome.
Radiologic Findings
The advent of MRI has provided for continuing improvement in the radiologic assessment of intracranial optic glioma involvement when compared to CT. Because optic gliomas are normally low grade and comprised of bland astrocytic proliferations, they are classically nearly isodense to normal brain.41 CT contrast enhancement varies from none to moderate so that the detection of the tumor and its margins with this modality is limited.43 CT, however, does image the bony alterations induced by the tumor. Optic foraminal enlargement has been shown to be present in approximately 70% of cases.46 Bony remodeling of the anterior clinoid process and chiasmatic groove may enlarge and deform the fossa and optic canal, which results in the classical J-shaped deformity or variations thereof in the sella turcica.62 Similar bony changes, however, may be induced by pressure from posterior extension of intraorbital neurofibroma, perioptic extension of craniopharyngioma, or superior extension of pituitary adenoma.28
With the capacity to visualize posteriorly located gliomas that cannot be detected by CT, MRI has many advantages.63 This contrasts with anteriorly located tumors that are detected equally well with both methods.63 This may be partially attributable to the fact that MRI eliminates bony artifacts while enhancing the delineation of abnormalities in brain tissue, which makes MRI superior in evaluating the intracanalicular, chiasmal, optic tract, optic radiation, or suprasellar hypothalamic extension of the tumor.19,64,65,66 An additional benefit of MRI is the lack of ionizing radiation, especially in cases when serial scanning is necessary to detect the presence or progression of tumor.
Intracranial optic gliomas may demonstrate imaging characteristics, which when interpreted with clinical findings, may provide for a definitive diagnosis without biopsy.21,67 These characteristics were identified by Hoyt and associates37,43 using CT scanning. They were:
Tubular thickening of optic nerve(s) and chiasm
Suprasellar globular tumor extension with contiguous optic nerve expansion
Suprasellar globular tumor extension with optic tract involvement
Tumors in the suprasellar area without these characteristics may necessitate biopsy to establish the diagnosis. The differential diagnosis of optic nerve glioma imaging includes germinoma and choristoma that may appear intrinsic to the visual pathway.21 Sellar enlargement may be created by craniopharyngioma and pituitary adenoma, although typically do not appear to be intrinsic to the visual pathway. Enlargement and enhancement of the chiasm or leptomeninges is uncommon in glioma and is suspicious for an inflammatory etiology.68 Meningiomas occur in children, but are less common. Unlike gliomas, they enhance strongly on MRI with gadolinium.
Optic nerve glioma in NF-1
NF-1 is one of the most common autosomal dominant disorders with a prevalence of 1:3500.69 Half of the patients affected by NF-1 are sporadic cases of the disease and represent new mutations of the gene located at the 17q11.2 locus.70 This gene, termed the NF-1 gene, is one of the largest human genes and has one of the highest mutations rates.71 Composed of 60 exons and spanning more than 300 kb of genomic DNA, the gene undergoes a spectrum of mutations, each of which may affect any of a large number of coding exons, which results in differently truncated or altered proteins.72 In NF-1-associated optic gliomas, most mutations affect the first exons of the gene, particularly exon 7.71 This mutational heterogeneity may underlie the variable course of patients with these tumors.
Considerable phenotypic variability has necessitated consensus NIH diagnostic clinical criteria, first defined in 198773 and later revised and updated.69,74 According to these criteria, the diagnosis is met if at least two of the following are present: six or more café au lait spots; two or more neurofibromas; one plexiform neurofibroma; axillary or inguinal freckling; optic glioma; two or more Lisch nodules; a distinctive osseous lesions such as sphenoid dysplasia; or a first-degree relative with NF-1.69,75 The lesions in NF-1 represent hamartomas and neoplastic proliferations of cells of neural crest and neuronal origin.76 Of these, plexiform neurofibroma, Lisch nodules, and optic gliomas are the most common ophthalmic manifestations. Other reported ophthalmic manifestations include colobomas, microphthalmos, or macrophthalmos.77
The incidence of NF-1 in patients with optic pathway gliomas has varied in reports from 10% to 70%9,17,24,78,79,80,81,82,83 with an overall incidence of 29% in published cases.10 Thus, the detection of an optic pathway glioma should prompt clinical evaluation for NF-1. However, optic pathway gliomas are not strictly associated with NF-1 because they have been reported to be present in patients with NF-2.84,85 Conversely, NF-1 patients should be evaluated for optic pathway disease, because an optic pathway tumor incidence has been reported to be 15% in NF-1 children without visual complaints.86 Listernick and co-workers86,87 stressed the significance of regular ophthalmologic examinations in children with NF-1, because normal neuroimaging of infants and young children with this disorder does not preclude the development of an optic pathway glioma later in childhood.88,89 Even though some investigators have demonstrated optic pathway gliomas to be present in asymptomatic children using imaging,90,91 it was not recommended by the NIH consensus development conference for routine screening.75
CT and MRI imaging of patients with NF-1 frequently reveals more extensive gliomas of the visual pathway than seen in patients without NF-1.63,92 Because multiple lesions are often seen in NF-1,93,94,95 the presence of bilateral optic gliomas strongly favors the diagnosis of NF-1.63,92 The more extensive tumor involvement in NF-1 may be associated with more severe visual impairment at the time of diagnosis, but is not predictive of a worse prognosis because progressive visual dysfunction appears to be less frequent and severe in NF-1-associated gliomas.92 In NF-1, greater degrees of astrocytic proliferation may result in more pronounced elongation of the affected optic nerve, accentuating optic nerve S-shaped deformity and kinking, whereas greater degrees of meningothelial hyperplasia often cause tubular thickening of the optic nerve, which causes a high MRI T2 signal.66,82 Additional intracranial abnormalities in NF-1 patients include aqueductal stenosis, macrocephaly, and nonspecific T2 signals of the basal ganglia, internal capsule, midbrain, cerebellum, and subcortical white matter.76,91,96
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


