 Optic Nerve, Meningeal, and Other Neural Tumors
Optic Nerve, Meningeal, and Other Neural TumorsOptic Nerve Juvenile Pilocytic Astrocytoma (Optic Nerve Glioma)
General Considerations
Juvenile pilocytic astrocytoma (JPA; optic nerve glioma) is a well known and important optic nerve and brain tumor of childhood (1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20). It represents 2% to 5% of brain tumors in children and accounts for about 1% to 2% of all orbital tumors (2,3,4,8). In the authors’ series of biopsy-proven cases, there were 4 cases among 645 biopsied orbital lesions (2). In the authors’ subsequent review of biopsy-proven childhood orbital tumors, it accounted for 5 of 250 biopsies (2%) (3). In the authors’ more recent clinical series of 1,264 consecutive space-occupying orbital lesions, the 48 JPAs accounted for 46% of the 105 optic nerve lesions and for 4% of all orbital lesions (4).
JPA has a definite association with neurofibromatosis type 1 (NF1) (1,19,20). Although figures vary from series to series, we believe that .50% of patients with JPA of the optic nerve have evidence of NF1. It is generally believed that JPA associated with NF1 is less aggressive and can regress (19). In contrast sporadic JPA has an earlier onset and more severe clinical course than the nonsporadic type associated with NF1 (8). JPA of the optic nerve has also been seen in association with von Hippel-Lindau syndrome, but may be coincidental (11). There is a definite predilection for females (13).
Clinical Features
Some JPAs of the optic nerve are asymptomatic and are discovered on routine orbital magnetic resonance imaging (MRI) screening in a patient with NF1. Usually, however, JPA is diagnosed in the first or second decade of life when the child presents with progressive visual loss and axial proptosis. It occasionally becomes clinically apparent in adulthood (6). Because there is a high incidence of NF1 associated with this tumor, the affected patient should be evaluated for cutaneous pigmented macules (café-au-lait spots), iris Lisch nodules, and other stigmata of NF1. In the early stages, fundus examination may show a swollen optic disc; later it may become pale with the appearance of a retinochoroidal shunt vessel on the margin of the disc (1). The chronic disc edema can rarely induce a choroidal neovascular membrane in the posterior pole of the eye (18).
JPA located more posteriorly in the intracranial portion of optic nerve, chiasm, or hypothalamus often produces visual loss, strabismus, or nystagmus. Proptosis is not generally present unless the lesion extends anteriorly into the orbit. JPA of the optic nerve can occasionally undergo spontaneous regression, in both sporadic cases and those associated with NF1 (12,17).
Diagnostic Approaches
Computed tomography (CT) and MRI of optic nerve JPA typically shows an ovoid mass corresponding to an enlarged optic nerve. A characteristic kink in the midportion of the tumor is often present. Extension into the optic foramen to the chiasm and brain is common and is best seen with gadolinium-enhanced MRI. The extent of the tumor, as determined by MRI findings, can help to predict visual loss, with postchiasmal lesions causing greatest visual impairment.
Pathology
Gross pathology shows an optic nerve mass that is typically surrounded by the dura mater. Histopathologically, JPA has typical histopathologic characteristics (1,3). It is composed of a benign proliferation of pilocytic (hairlike) astrocytes, sometimes with areas of mucinous degeneration and hemorrhage. More round cells are often present. The astrocytic cell processes sometimes have eosinophilic, cylindrical swellings, called Rosenthal fibers (1,3).
Management and Prognosis
The management of JPA of the optic nerve is complex and controversial. Because the lesion is benign, we generally prefer to be as conservative as possible. Biopsy is rarely necessary today, because the diagnosis can usually be established based on the characteristic radiographic findings. Asymptomatic lesions are often followed initially without treatment and many remain stable for long periods of time (9). Serial examinations should be done for pupillary reaction, visual acuity, visual fields, and color vision of both eyes. Most tumors confined to the orbit remain relatively stable, but slow growth can occasionally occur. If the patient is blind and has unacceptable proptosis of the affected eye, then complete removal of the mass by lateral orbitotomy is warranted. For those that extend to the orbital apex or more posteriorly, a neurosurgical approach is necessary. It is usually not necessary to remove the blind eye in such cases. Progressive lesions that involve the chiasm and brain are sometimes fatal and may require radiotherapy. The role of chemotherapy is uncertain, but results from early studies have been encouraging. When the tumor is initially confined to the optic nerve, the mortality is ,5%. When the hypothalamus is involved, mortality rises to .50% (9).
Selected References
1. Shields JA. Juvenile pilocytic astrocytoma. In: Shields JA, ed. Diagnosis and Management of Orbital Tumors. Philadelphia: WB Saunders; 1989:170-179.
2. Shields JA, Bakewell B, Augsburger DG, et al. Classification and incidence of space-occupying lesions of the orbit. A survey of 645 biopsies. Arch Ophthalmol 1984;102:1606-1611.
3. Shields JA, Bakewell B, Augsburger DG, et al. Space-occupying orbital masses in children. A review of 250 consecutive biopsies. Ophthalmology 1986;93:379-384.
4. Shields JA, Shields CL, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions: The 2002 Montgomery Lecture, part 1. Ophthalmology 2004;111:997-1008.
5. DePotter P, Shields JA, Shields CL. Optic nerve and meningeal tumors. In: DePotter P, Shields JA, Shields CL, eds. MRI of the Eye and Orbit. Philadelphia: JB Lippincott; 1994:193-201.
6. Wulc AE, Bergin DJ, Barnes D, et al. Orbital optic nerve glioma in adult life. Arch Ophthalmol 1989;107:1013-1016.
7. King A, Listernick R, Charrow J, et al. Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am J Med Genet 2003;122:95-99.
8. Czyzyk E, Jozwiak S, Roszkowski M, et al. Optic pathway gliomas in children with and without neurofibromatosis. J Child Neurol 2003;18:471-478.
9. Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol 1994;38:427-452.
10. Khafaga Y, Hassounah M, Kandil A, et al. Optic gliomas: a retrospective analysis of 50 cases. Int J Radiat Oncol Biol Phys 2003;13:807-812.
11. Nau JA, Shields CL, Shields JA, et al. Optic nerve glioma in a patient with von Hippel-Lindau syndrome. J Pediatr Ophthalmol Strabismus 2003;40:57-58.
12. Parsa CF, Hoyt CS, Lesser RL, et al. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol 2001;119:516-529.
13. Gayre GS, Scott IU, Feuer W, et al. Long-term visual outcome in patients with anterior visual pathway gliomas. J Neuroophthalmol 2001;21:1-7.
14. Balcer LJ, Liu GT, Heller G, et al. Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: relation to tumor location by magnetic resonance imaging. Am J Ophthalmol 2001;131:442-445.
15. Thiagalingam S, Flaherty M, Billson F, et al. Neurofibromatosis type 1 and optic pathway gliomas: follow-up of 54 patients. Ophthalmology 2004;111:568-577.
16. Jakobiec FA, Depot MJ, Kennerdell JS, et al. Combined clinical and computed tomographic diagnosis of orbital glioma and meningioma. Ophthalmology 1984;91:137-155.
17. Schmandt SM, Packer RJ, Vezina LG, et al. Spontaneous regression of low-grade astrocytomas in childhood. Pediatr Neurosurg 2000;32:132-136.
18. Shields JA, Shields CL, De Potter P, et al. Choroidal neovascular membrane as a feature of optic nerve glioma. Retina 1997;17:349-350.
19. Stern J, Jakobiec FA, Housepian EM. The architecture of optic nerve gliomas with and without neurofibromatosis. Arch Ophthalmol 1980;98:505-511.
20. Hoyt WF, Baghdassarian SA. Optic glioma of childhood, natural history and rationale for conservative management. Br J Ophthalmol 1969;53:793-798.
Optic Nerve Juvenile Pilocytic Astrocytoma (Glioma)
Juvenile pilocytic astrocytoma of the optic nerve has typical clinical and computed tomography features. When it is confined to the orbit and produces a blind eye with progressive irreversible proptosis, the tumor can be removed by a lateral orbitotomy. A clinicopathologic correlation is shown.
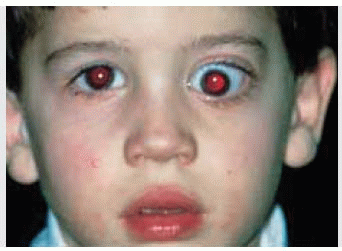
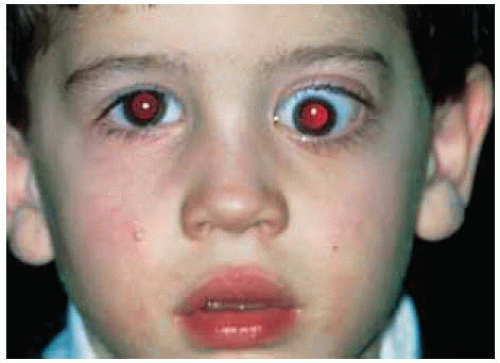 Figure 30.1. Axial proptosis of the left eye in a 4-year-old boy. The proptosis had been progressive for more than a year. |
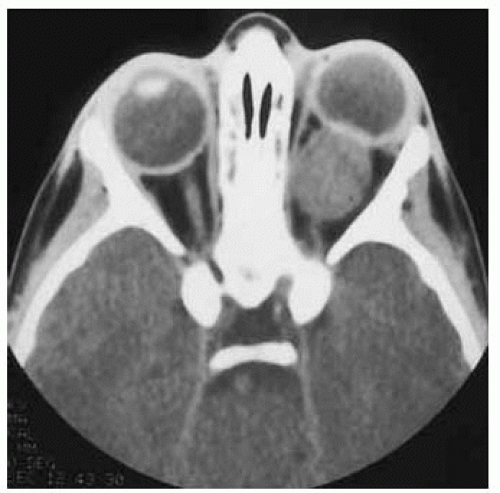 Figure 30.2. Axial computed tomography of patient shown in Figure 30.1 revealing a characteristic, well-defined, ovoid mass affecting the optic nerve. The lesion had shown considerable enlargement compared to a CT done 1 year earlier. |
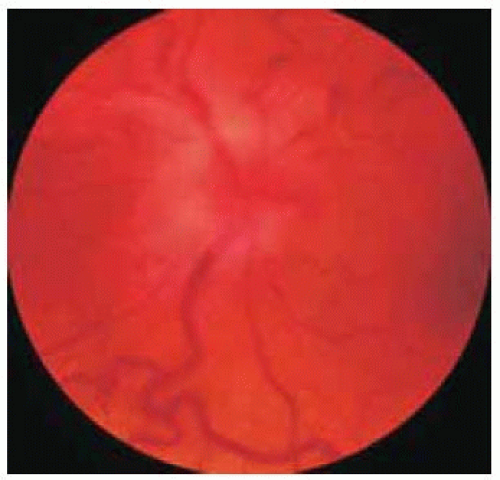 Figure 30.3. Marked swelling of the optic disc of the left eye. |
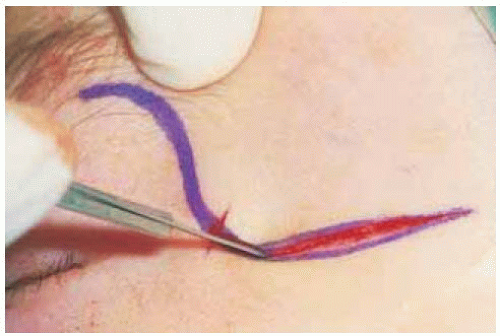 Figure 30.4. The proptosis continued to increase to an unacceptable degree, and surgical excision was elected. Shown is the planned skin incision for a superolateral orbitotomy. |
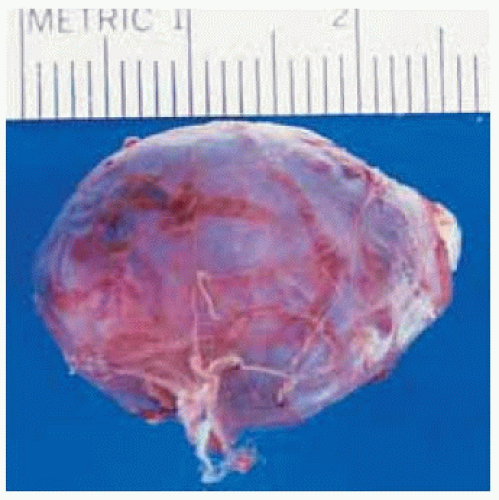 Figure 30.5. Gross appearance of the well-circumscribed mass immediately after surgical removal. |
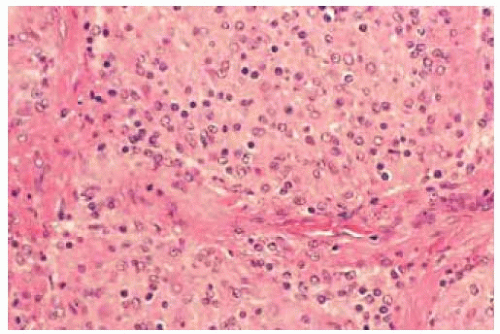 Figure 30.6. Histopathology showing closely compact astrocytes with some round nuclei comprising the tumor. (Hematoxylin-eosin 100.) |
Optic Nerve Juvenile Pilocytic Astrocytoma (Glioma): Magnetic Resonance Imaging
Juvenile pilocytic astrocytoma of optic nerve can be diagnosed with either computed tomography or magnetic resonance imaging because of its typical imaging features. However, MRI with gadolinium enhancement and fat suppression is the best method for determining whether there is subtle extension of the tumor into the optic canal and chiasmal area. Two examples of MRI are shown.
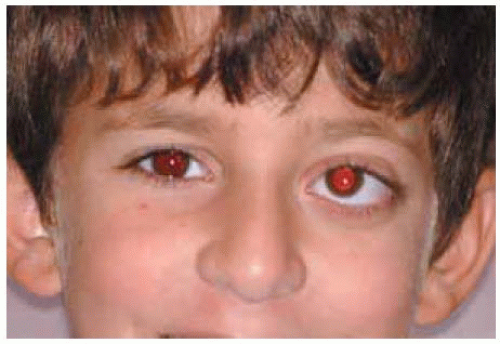 Figure 30.7. Proptosis and upward displacement of right globe in a 12-year-old boy. |
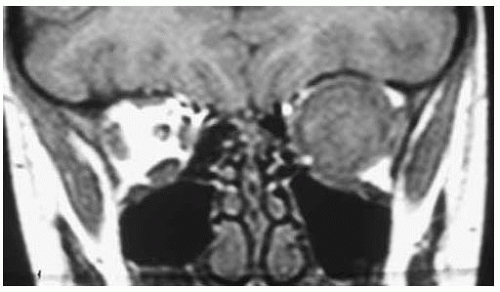 Figure 30.8. Coronal magnetic resonance imaging in T1-weighted image of patient shown in Figure 30.7. Note the large round tumor with its center in the location of the optic nerve. |
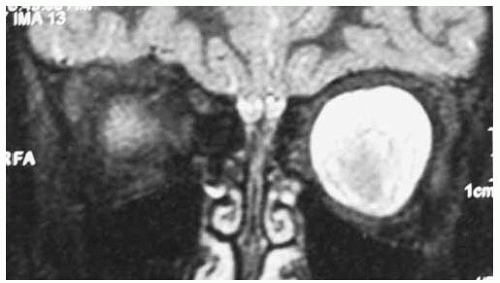 Figure 30.9. Coronal magnetic resonance imaging in T1-weighted image with gadolinium enhancement and fat suppression of patient shown in Figure 30.7. The geographic central area could represent the tumor and the surrounding hyperintense area could represent arachnoidal proliferation around the central tumor. |
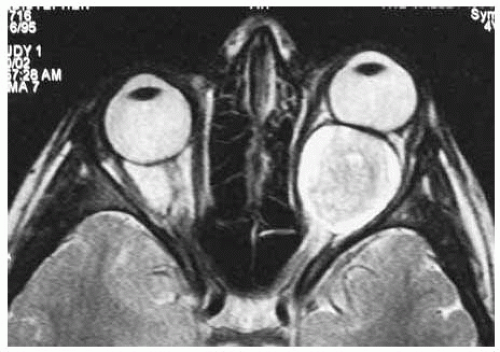 Figure 30.10. Axial magnetic resonance imaging in T2-weighted image of same patient, showing similar findings. |
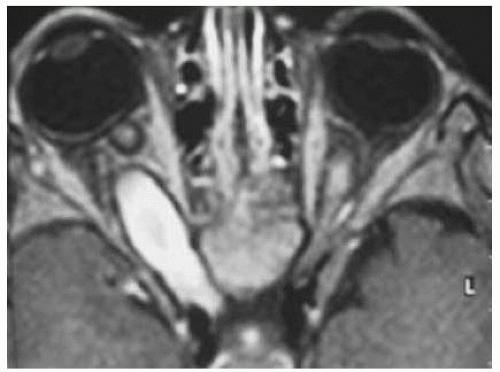 Figure 30.11. Another patient shown with axial magnetic resonance imaging in T1-weighted image with gadolinium enhancement and fat suppression of a posterior orbital juvenile pilocytic astrocytoma with extension into the right optic canal and optic tract. Note the hyperintense fusiform mass and dark rim around the mass that represents cerebrospinal fluid. |
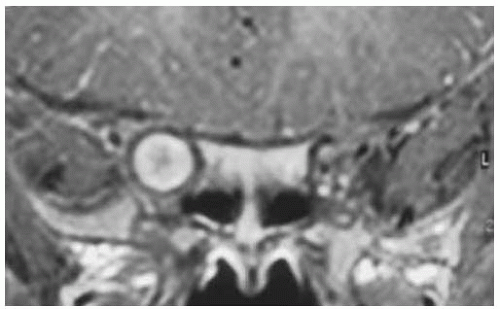 Figure 30.12. Coronal magnetic resonance imaging in T1-weighted image with gadolinium enhancement and fat suppression in same patient. This shows extension of the juvenile pilocytic astrocytoma almost to the chiasm. |
Optic Nerve Juvenile Pilocytic Astrocytoma (Glioma)
Juvenile pilocytic astrocytoma of the optic nerve has characteristic features with magnetic resonance imaging. When the proptosis is more advanced, the proptosis converts from an axial direction to a down-and-out direction, conforming to the contour of the bony orbit. Two related cases are illustrated.
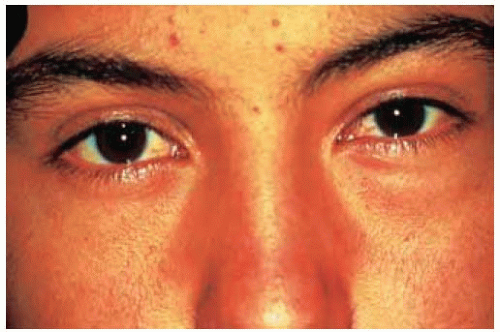 Figure 30.13. Proptosis of left eye in a 15-year-old boy. |
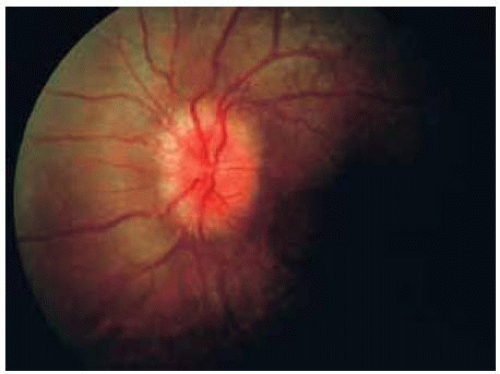 Figure 30.14. Hyperemia and edema of optic disc in patient shown in Figure 30.13. |
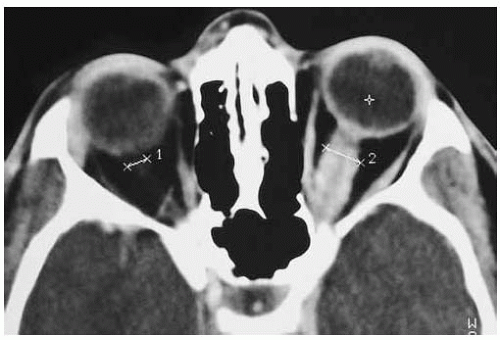 Figure 30.15. Axial computed tomography of patient shown in Figure 30.13. Note the fusiform lesion of the optic nerve. |
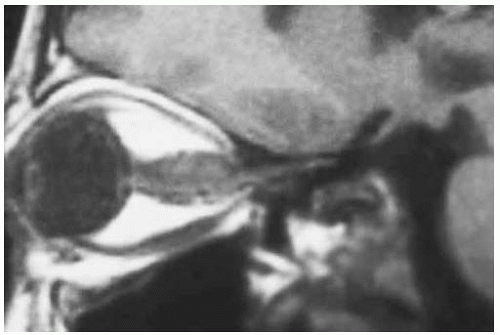 Figure 30.16. Sagittal magnetic resonance imaging in T1-weighted image showing same lesion depicted in Figure 30.15. |
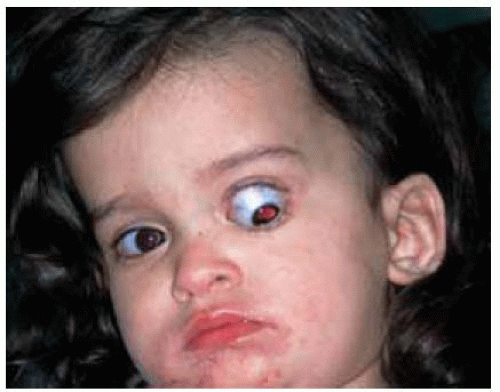 Figure 30.17. Proptosis and dow nward displacement of the left eye secondary to a juvenile pilocytic astrocytoma in a 2-year-old girl. The eye was blind and the progressive proptosis prompted surgical removal of the tumor. |
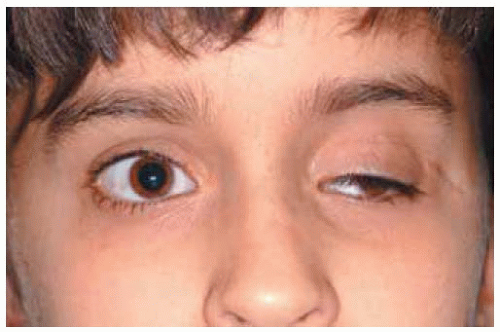 Figure 30.18. Appearance of child shown in Figure 30.17 after removal of the tumor. There is no more proptosis and the blepharoptosis and exotropia are to be corrected in the future. |
Optic Nerve Juvenile Pilocytic Astrocytoma (Glioma): Fundus Changes
The most common fundus changes with juvenile pilocytic astrocytoma of the optic nerve are a swollen optic disc followed by a retinochoroidal shunt vessel and pallor of the optic disc. The venous stasis secondary to optic disc involvement can prompt development of a juxtapapillary choroidal neovascular membrane.
Shields JA, Shields CL, De Potter, et al. Choroidal neovascular membrane as a presenting feature of optic nerve glioma. Retina 1997;17:349-350.
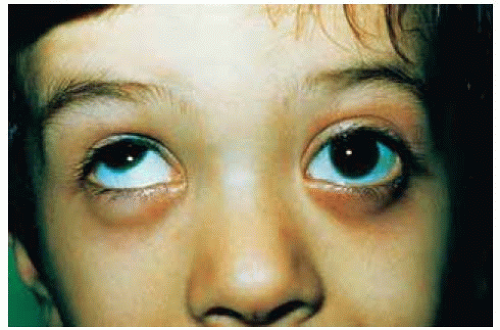 Figure 30.19. Proptosis of left eye and inability of upgaze secondary to a juvenile pilocytic astrocytoma of the optic nerve in a 6-year-old boy. |
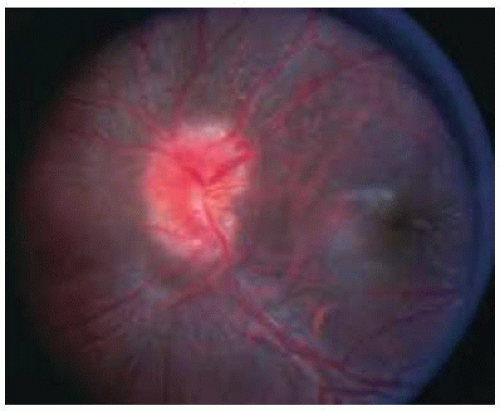 Figure 30.20. Swelling and hyperemia of the optic disc in the child shown in Figure 30.19. Note the retinochoroidal shunt vessel on superotemporal margin of optic disc. |
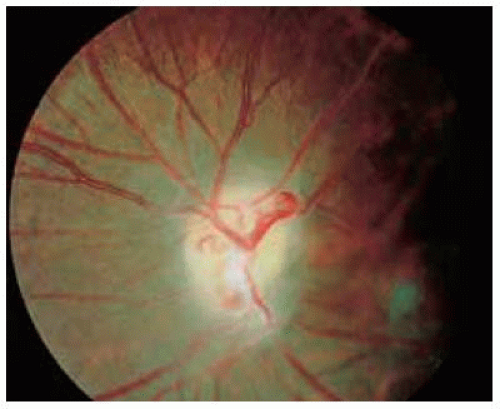 Figure 30.21. Appearance of same optic disc 4 years later. Note the optic disc pallor and distinct retinochoroidal shunt vessel. |
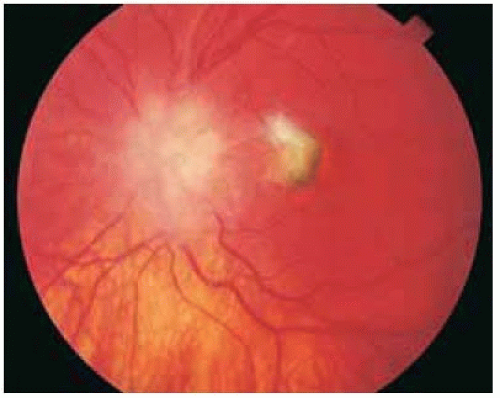 Figure 30.22. Choroidal neovascular membrane temporal to chronically swollen optic disc in a 16-year-old girl. The cause of the optic disc changes and neovascular membrane was not initially determined. |
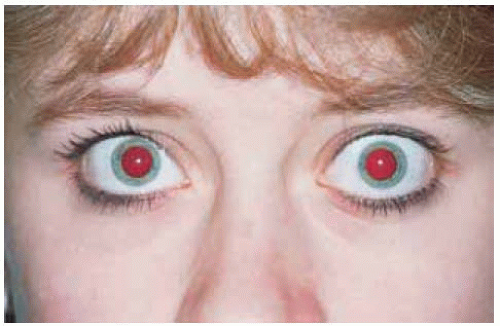 Figure 30.23. A few weeks later the patient shown in Figure 30.22 was realized to have proptosis of the left eye, which prompted an orbital computed tomography and referral. |
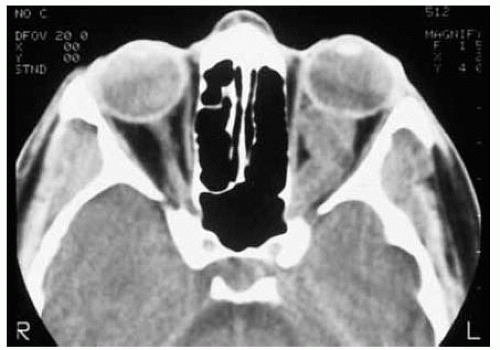 Figure 30.24. Axial computed tomography of patient shown in Figure 30.23 demonstrating the characteristic findings of a juvenile pilocytic astrocytoma of the optic nerve. Note the characteristic kink in the midportion of the nerve nasally. |
Optic Nerve Malignant Astrocytom
General Considerations
The benign pilocytic astrocytoma usually has its clinical onset in childhood. There is also a rare malignant form of optic nerve astrocytoma that occurs in adulthood and that is not usually associated with neurofibromatosis (1,2,3,4,5,6,7,8,9,10,11,12,13). Its clinical features, histopathology, and clinical course are very different from the juvenile pilocytic astrocytoma. Fewer than 50 cases have been published (13). Although it is generally considered to be rare, it is possible that many such cases have gone unreported because the patients usually die rapidly with a “brain tumor” and the precise diagnosis may not always be established.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


