Fig. 7.1
Longitudinal section of optic nerve. The lamina cribrosa sieve-like partition is to the right of the image. The central retinal artery and vein are seen at the left of the image
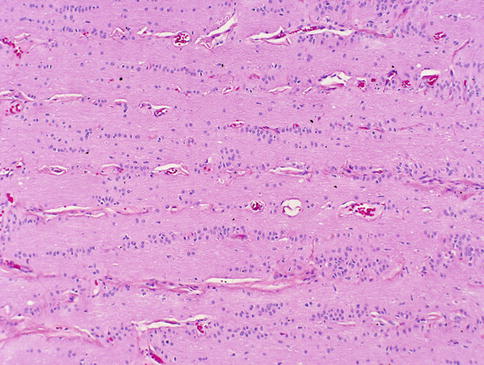
Fig. 7.2
Higher magnification of this figure shows the orderly columnar arrangement of optic nerve glia
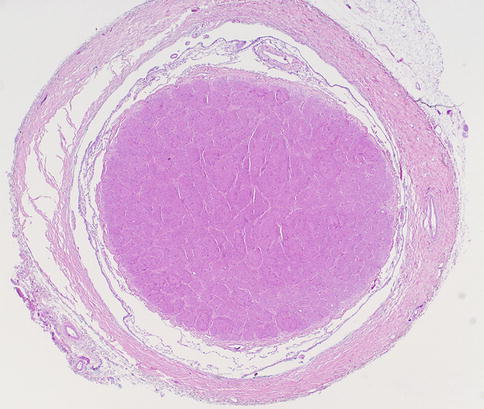
Fig. 7.3
Normal optic nerve in cross section surrounded by meninges
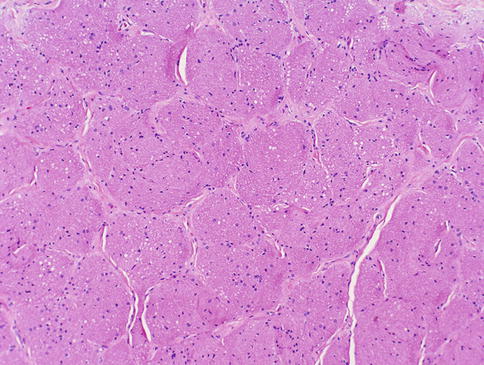
Fig. 7.4
Higher magnification of this figure shows the compartmentalized appearance of the nerve in cross section
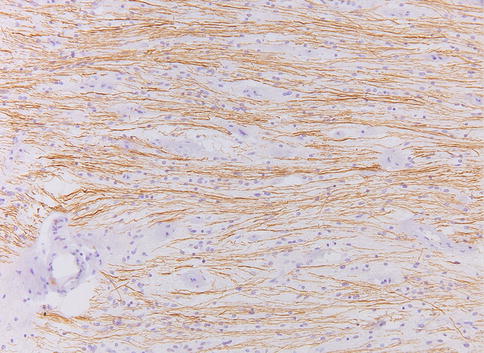
Fig. 7.5
Normal optic nerve. Neurofilament axonal immunohistochemical stain of a longitudinal section of the nerve
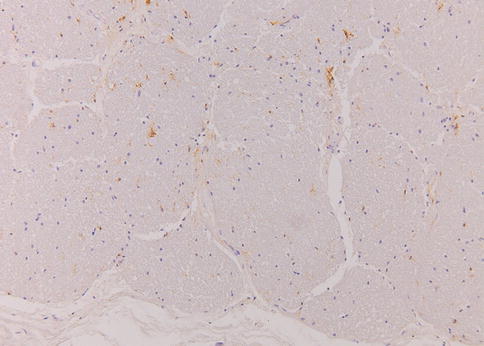
Fig. 7.6
Normal optic nerve. HLA-DR immunohistochemical stain demonstrating scattered microglial cells in the optic nerve. The relative light staining indicates the microglial cells are not activated
The optic nerve is subject to a wide range of physiological and mechanical pathologies. The normal clinical appearance of the optic nerve head (Fig. 7.7a) must be contrasted with the appearance of a swollen nerve head (Fig. 7.7b). Papilledema or swelling of the tissues of the optic nerve head may result from a variety of causes, including increased intracranial pressure, increased intraorbital pressure, systemic hypertension, and hypercapnia. Papilledema can often be diagnosed during ophthalmological examination, and a careful search for its etiology should ensue. Histological examination may disclose edema, hemorrhages, exudate, and axonal swelling. Prolonged swelling may result in a secondary glial proliferation [1]. Optic nerve atrophy and cupping of the disk are well-recognized effects of increased ocular pressure (glaucoma) and the degenerative sequelae of retinal ganglion cell loss. The nerve is also affected by a range of metabolic, vascular, and nutritional anomalies, many of which will only be apparent if the nerve can be examined postmortem.
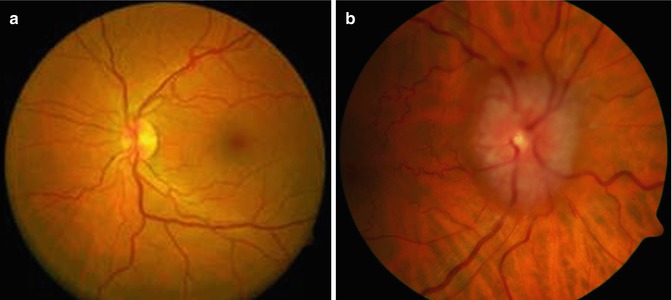
Fig. 7.7
(a) Clinical image of normal fundus. (b) Papilledema: clinical image of the fundus showing marked swelling of the optic nerve head
7.2 Embryology, Anatomy, and Development
The development of the eye is an incredibly complex sequence of events. Three primary bulges appear in the future brain region of the neural tube: the forebrain (prosencephalon), the midbrain (mesencephalon), and the hindbrain (rhombencephalon). A secondary bulge, the optic vesicle, extends from each side of the forebrain and differentiates to form an optic cup, an optic stalk, and eventually an optic nerve and different components of the eye.
For the optic nerve, there are a few crucial processes, but since development is an orderly sequence, any disruptions, even in “distant” parts of the eye, may influence formation of apparently non-related parts or segments. The first important process is the formation of the optic cup itself and the thinning of the connection to the neural tube, i.e., the formation of the optic stalk, at approximately 1 month of gestation. The neuroepithelium of the optic stalk will act as a supportive layer for the axons of the ganglion cells (developing in the retina) during the 2nd month, forming the optic nerve proper. The second important process is the formation of the embryonic or optic fissure; this will make place for the hyaloid vascular system that will vascularize the inner segments of the eye. The fissure will eventually envelop the hyaloid artery and will close. The closure of the fissure occurs in the 2nd month. From the 4th month the hyaloid vessel system starts to regress, but the more proximal parts will remain, forming the central retinal artery and vein, and later the vascularization of the retina. Myelination of the fibers of the optic nerve will start in the third trimester and will be completed only after birth.
7.3 Congenital Abnormalities
Congenital or developmental disorders which affect the optic nerve are more likely to be recognized in postmortem examinations than in surgical specimens. Optic nerve aplasia and optic nerve hypoplasia likely are part of a spectrum of aberrant development and ocular embryogenesis. True optic nerve aplasia occurs when the optic nerve fails to develop, either because the retinal ganglion cells do not develop or they fail to form nerve fibers. In optic nerve hypoplasia, the nerve has fewer nerve fibers than normal, and there are a diminished number of retinal ganglion cells. Optic nerve aplasia and hypoplasia may either occur unilaterally or bilaterally. Some other developmental anomalies of the optic nerve include megalopapilla, tilted disks, optic nerve head pits, Aicardi syndrome, peripapillary staphyloma, myelinated nerve fibers, optic nerve drusen, and optic disk pigmentation [1]. In the morning glory syndrome, a funnel-shaped optic nerve disk is surrounded by chorioretinal pigmentation, and centrally the disk appears to contain Bergmeister’s papilla representing persistence of the hyaloid system.
Colobomas of the optic disk occur when there is incomplete closure of the posterior portion of the fetal fissure. Colobomas can affect all parts of the globe, including the optic nerve, and they may be limited to the disk or may be associated with colobomas of the choroid, ciliary body, iris, retina, or sclera. A known association of coloboma is a microphthalmic eye with choristomatous malformation (microphthalmos with cyst).
Mitochondrial disease also can affect the optic nerve, the best example being Leber’s hereditary optic atrophy [4]. The histology is characterized by loss of axons and myelin, sometimes with a variable degree of inflammation, with macrophages clearing myelin debris, and some T lymphocytes. In this, the pathology can show similarities with that of multiple sclerosis [5].
7.4 Inflammation and Ischemia
7.4.1 Optic Neuritis
Optic neuritis is an inflammation of the optic nerve that has a variety of causes, including infectious, ischemic, and (autoimmune) demyelinating. Most of these conditions represent secondary involvement of the optic nerve. Clinically, almost invariably, inflammation of the nerve causes severe visual impairment.
7.4.1.1 Infection
Almost all infections of the optic nerve are secondary to infections elsewhere, reflecting the well-protected position of the nerve. However, intraocular infections, such as corneal ulcerations, endophthalmitis, and chorioretinitis, can spread to involve the nerve. Likewise, infections in the orbit themselves not infrequently originating from the sinonasal compartment (zygomycosis) [6] or from the cranial vault (meningitis) can involve the optic nerve by direct spread. More rarely, more distant infections can result in secondary optic nerve inflammation, e.g., bacterial endocarditis, tuberculous infections, syphilis, and others. All of these have become rarities, especially in the developed countries. However, in patients with compromised immune systems, even in the developed countries, rhino-cerebro-orbital infections are seen with some frequency, and it is necessary to realize that immune dysfunctions can be obvious (transplantation, AIDS [acquired immune deficiency syndrome] patients, and chemotherapy treatment) but also can be less so obvious (diabetes mellitus) [7, 8] (Fig. 7.8).
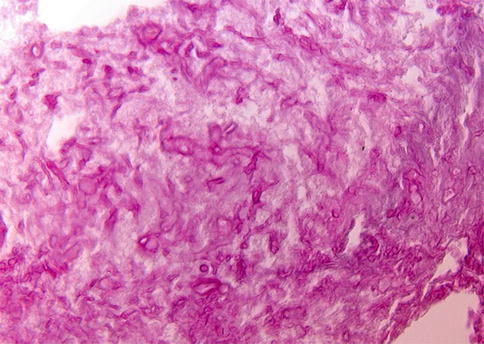
Fig. 7.8
Mucormycosis/zygomycosis. Periodic acid-Schiff (PAS)-stained orbital biopsy section in a patient with mucormycosis (the patient was diabetic). A large portion of the material was necrotic, due to vascular occlusion by the fungus. The characteristic broad and irregular hyphae can be seen, branching at right angles
Symptoms in individuals with inflammatory or infectious pathologies are variable degrees of visual impairment, and, of course, also the signs and symptoms of the underlying condition.
The pathology is highly inflammatory, with infiltration with many inflammatory cells, with the organism responsible determining the nature of the infiltrate. In some cases, it will be possible to demonstrate the offending organism with appropriate histochemical or immunohistochemical stains or molecular techniques (polymerase chain reaction). Fungal infections can show vasoinvasive properties that cause infarction and necrosis, contributing to optic nerve damage [9, 10]. This is the prime reason that in such cases surgical treatment is mandatory, as fungicidal antibiotics alone will not cure these patients.
7.4.1.2 Inflammation Secondary to Vascular Disease and Ischemia
The optic nerve can also be affected by ischemic changes secondary to vascular disorders including occlusion of the internal carotid artery, occlusion of the central retinal artery and vein, occlusion of the ophthalmic artery, carotid-cavernous sinus fistula, and vasculitis.
Vasculitis can be a local or systemic process, and this varied group of disorders can in principle affect any part of the body. Giant cell arteritis (also referred to as temporal arteritis) deserves mention as it not infrequently can lead to visual disturbances. It is a poorly understood condition in which there is chronic, often (partly) granulomatous mural inflammation of the somewhat larger arteries. The giant cells seem to focus on the internal elastic lamina, but they are not always found when an affected artery is biopsied (almost always the temporal artery) [11, 12] (Fig. 7.9). In such instances, the inflammation is usually composed of lymphocytes and plasma cells, and the designation giant cell arteritis is somewhat of a misnomer. When the inflammatory infiltrate consists of eosinophils, the possibility of Churg-Strauss syndrome should be considered, which may rarely involve the temporal artery. The pathology of the vasculitides, including the temporal artery, is known to be patchy and sometimes referred to as “skip lesions.” It is therefore recommended to submit a segment of artery at least two centimeters in length. The vessel should be placed on end, serially sectioned, submitted entirely, and multiple step sections submitted through the paraffin block; this will give the best chance to see a focal area of inflammation. Sometimes the inflammatory component might be quenched by preoperative steroid therapy, and there will scarring of the vessel wall consistent with a “healing” arteritis. A Masson trichrome histochemical stain can be useful to highlight the scarring.
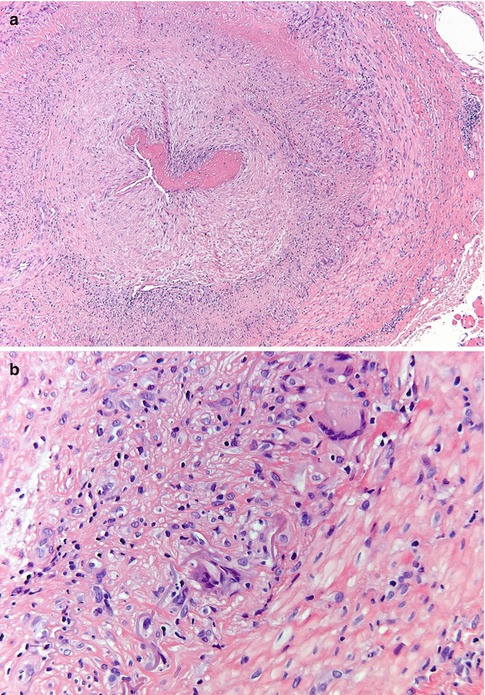
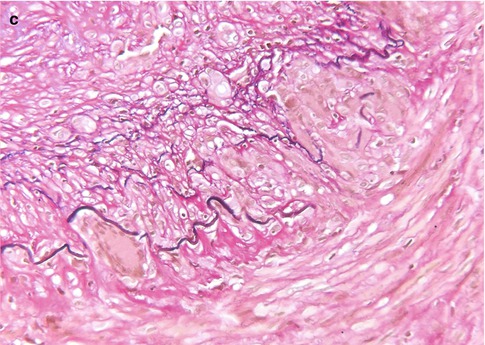
Fig. 7.9
Giant cell arteritis. (a) Temporal artery specimen showing transmural inflammation of the vessel wall (H&E). (b) A few multinucleated giant cells are identified and are centered on the internal elastic lamina (H&E). (c) Elastic stain showing fragmentation of the internal elastic lamina
Ischemia is the hallmark of two, probably related, conditions, whose pathophysiology is unclear: non-arteritic anterior ischemic optic neuropathy and neuroretinitis. They both target the anterior portion of the nerve and cause abrupt loss of visual acuity and visual field defects. The former is seen in a somewhat older age group. The histology is relatively noninflammatory, with edema [13].
7.4.1.3 Inflammation Secondary to Demyelinating and/or Autoimmune Disease
The primary category here is retrobulbar or optic neuritis in multiple sclerosis (MS) [14]. Clinically, this is characterized by a rapidly progressive loss of vision, especially central vision, disturbances in color vision, and sometimes pain [15]. In a few weeks vision recovers. The widely held concept that MS is an autoimmune disease in which myelin is attacked by the immune system is increasingly challenged; possibly the demyelination in MS is secondary [to axonal damage, for instance] and the inflammation in its turn is secondary to the demyelination. Whatever the sequence of events is, lesions in MS show influx of macrophages and a varying number of lymphocytes that are predominantly found in the perivascular spaces (i.e., outside the central nervous system tissue proper). The macrophages phagocytose myelin debris, turning into foam cells and clearing the way for remyelination. As the myelin in the center of a lesion is cleared, the macrophages also disappear, first leaving a rim of foamy macrophages around a hypocellular center, and eventually they disappear altogether, leaving a burnt-out, demyelinated, and hypocellular plaque [16, 17] (Fig. 7.10a–c). Remyelination ensues, but subsequent new attacks of demyelination exhaust the brain tissue’s regenerative capacity and eventually leave completely the above-described demyelinated, noninflammatory lesions or plaques. Most optic nerves from MS patients investigated involve postmortem material and will show these burnt-out, scar-like lesions. In rare instances a lesion will be found in which foamy macrophages can be found, with a little lymphocytic (perivascular) infiltrate. The lymphocytes are predominantly T lymphocytes. Invariably, there is astrogliosis, with hypertrophic astrocytes (i.e., astrocytes with abundant cytoplasm and cellular protrusions). The number of axons is variable; sometimes damage can be demonstrated by loss of axons and by axonal swellings, indicative of damage. In other cases changes are hardly noticeable (Fig. 7.10d).
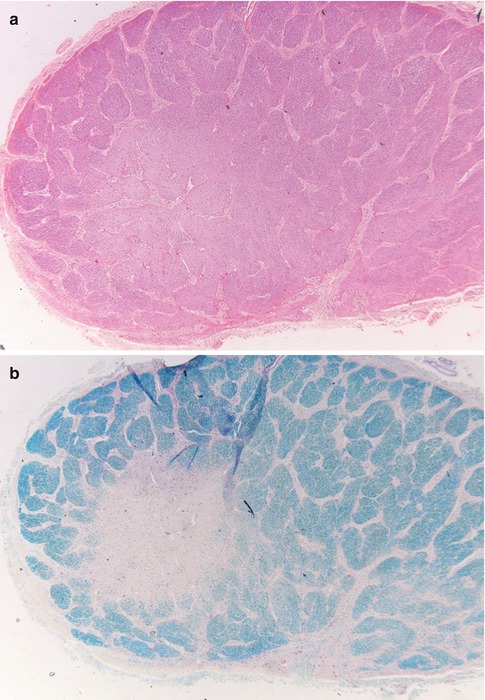
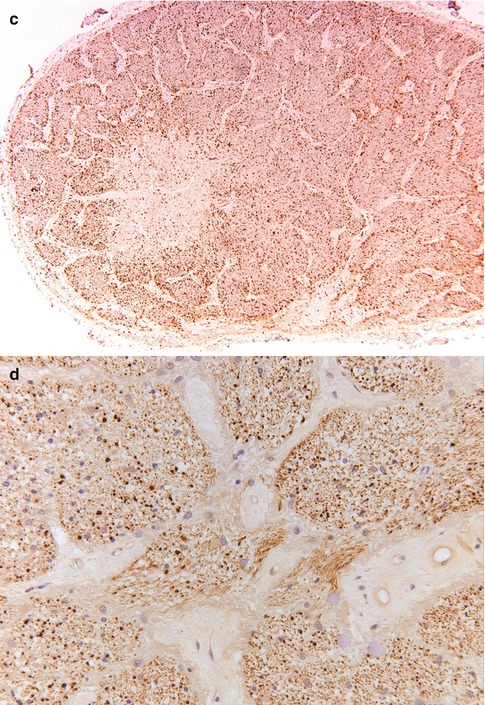
Fig. 7.10
Multiple sclerosis. (a) Optic nerve with a paracentral zone of pallor (H&E). (b) Luxol fast blue stain shows reduced myelin staining consistent with myelin loss in the nerve. (c) HLA-DR staining. The center of the lesion is less heavily infiltrated with microglial cells, whereas the rim shows increased numbers of activated microglia. (d) Neurofilament stain shows no clear difference areas in the number of axons between the demyelinated area and the myelinated area
Neuromyelitis optica (NMO) or Devic’s disease was once considered a potential variant of MS. However, NMO is now known to be caused by an autoimmune reaction to aquaporin 4, a water channel protein on astrocytes [18]. It is, therefore, a true autoimmune disease. Clinically, there is rapid loss of vision to complete blindness and paraplegia, caused by a destructive process in the spinal cord. The brain is typically not involved, although it may be involved in some cases. The characteristic abnormalities in the optic nerve are demyelination and the disappearance of astrocytes from the tissue. In the acute phase there are many inflammatory cells, including lymphocytes and foamy macrophages, but often also neutrophils.
A variety of drugs, metabolic diseases, and radiation therapy can also cause secondary inflammation of the optic nerve.
7.5 Injuries
External force or trauma to the optic nerve may result in mechanical injury including stretching, tearing, torsional stress, avulsion at the optic disk, avulsion at the optic canal, and chiasm. Penetrating injuries from foreign bodies or iatrogenic injections have been reported. Compression of the nerve may occur secondary to hemorrhage, inflammation, tumors such as meningiomas, and thickening of the extraocular muscles in cases of thyroid ophthalmopathy [1].
7.6 Degenerations
Degeneration of the optic nerve typically refers to optic atrophy, a clinical term describing optic pallor, avascularity, and cupping of the optic disk. Cavernous degeneration is a form of optic atrophy characterized by the formation of hyaluronic acid-filled cavernous spaces in the retrolaminar/anterior segment of the nerve. Hyaluronic acid is an acid mucopolysaccharide that is sensitive to hyaluronidase and is believed to be derived from the vitreous. Schnabel’s cavernous optic atrophy refers to the association of cavernous degeneration with glaucoma [1]. Alcian blue and colloidal iron stains can be useful in highlighting the mucopolysaccharide accumulations.
7.7 Optic Nerve Tumors
7.7.1 Introduction
Tumors of the optic nerve, optic chiasm, and optic nerve sheath comprise a narrow yet fascinating spectrum of lesions familiar to the ophthalmic pathologist and neuropathologist. These include optic nerve gliomas; meningiomas of the optic nerve sheath; melanocytomas and medulloepitheliomas of the optic nerve head; secondary tumors including metastases, retinoblastoma with nerve invasion, and retinoinvasive uveal melanoma; hemangiopericytoma and hemangioblastoma; and optic nerve choristoma. Finally, a few other rare reported tumors involving the nerve and its sheath will be mentioned.
A review of the early history of optic nerve gliomas can be found in the textbook of the late Sir W. Stewart Duke-Elder, with references about the earliest case reports dating back to the early nineteenth century [19]. The occurrence of an optic nerve glioma with von Recklinghausen neurofibromatosis was first noted by von Michel in 1873, and a significant association between these two entities was documented by Davis in 1940 [20]. In 1912, Hudson reviewed 182 optic nerve tumors, distinguished optic nerve gliomas from optic sheath meningiomas, and divided them into three classes: gliomatous (within the nerve), endotheliomatous, and fibromatous (associated with the nerve sheath) [21]. In 1922, Verhoeff noted that some tumors of the optic nerve were gliomas similar to those of the brain and had no relation to malignant retinal “gliomas” (retinoblastomas) [22]. Shortly thereafter, Martin and Cushing described seven gliomas involving the intracranial portion of the optic nerve and chiasm [23].
7.7.2 Optic Nerve Gliomas
Definition
Gliomas of the optic nerve are tumors derived from glial (astrocytic) cells within the optic nerve and chiasm. Optic nerve gliomas are traditionally thought of as pilocytic astrocytomas. However, low-grade (pilocytic astrocytoma, diffuse astrocytoma) and high-grade (glioblastoma) astrocytomas may involve the nerve, and therefore, an attempt to best grade and classify these tumors should be made. Correlation with clinical features, neuroimaging, and intraoperative findings is recommended in these cases.
Epidemiology
Optic nerve gliomas represent approximately 4 % of intracranial gliomas and 66 % of primary optic nerve tumors [24–27]. Approximately 90 % are diagnosed within the first two decades of life. The median age is 7 years and ranges from birth to the eighth decade. The female-to-male ratio is approximately 3:2, and both genders appear to be equally affected when the neoplasm involves the optic chiasm.
Etiology
Optic nerve gliomas derive from glial cells within the optic nerve. While the term “glioma” comprises astrocytic, oligodendroglial, and ependymal neoplasms, ependymal tumors have not been reported in the optic nerve, and only rare putative cases of oligodendroglioma are in the literature. Glioneuronal tumors (ganglioglioma) are exceptionally rare in the optic nerve.
Clinical Features
Individuals with optic nerve gliomas may present clinically with a wide spectrum of ophthalmic abnormalities including visual impairment, optic atrophy, and proptosis [28–31]. Malignant gliomas of the anterior visual pathway are a clinical entity distinct from benign gliomas of childhood and they typically present with unilateral or bilateral visual loss and subsequent blindness [32]. The optic disk may initially appear normal but eventually evolves into papilledema or optic atrophy. These aggressive tumors can spread via the optic radiations, leptomeninges, or cerebrospinal fluid both intracranially and to the spinal neuraxis [33, 34]. Rare glioblastomas of the optic nerve have been reported in childhood, either arising de novo [35], from malignant transformation of a pilocytic astrocytoma [36], or secondary to radiation therapy [37, 38].
Macroscopy
Optic nerve pilocytic astrocytomas often exhibit a smooth, fusiform, intradural enlargement of the nerve and acquire an hourglass shape when they spread through the optic foramen. They are white to tan in color and often contain abundant mucous imparting a soft gelatinous consistency. Small cystic spaces and foci of hemorrhage are sometimes seen [39, 40]. Extensive hemorrhage within a cyst resulting in rapid proptosis is rare [41]. Two architectural forms can be seen at the gross and microscopic level. One is a diffuse expansion of the optic nerve without extensive subarachnoid spread, and the other has a prominent infiltration of the subarachnoid space causing a rim of tumor around a mildly involved nerve [26, 42, 43]. The ability to invade the subarachnoid space and exhibit leptomeningeal dissemination and metastasis confirms that optic nerve pilocytic astrocytomas are true neoplasms rather than hamartomas [26, 42, 44].
Primary gliomas of the optic nerve head may also involve the adjacent retina [45]. Aside from gliomas that arise in the optic nerve, others may infiltrate the nerve after originating in the optic chiasm, floor of the third ventricle, thalamus, or hypothalamus. Sometimes it may be difficult, if not impossible, to determine the exact site of tumor origin among these sites radiographically, intraoperatively, or at postmortem examination [45, 46].
Histopathology
Pilocytic astrocytomas may expand the septa of the optic nerve (Fig. 7.11) or exhibit a biphasic growth pattern. Malignant astrocytomas also expand the septa and must be considered in the differential diagnosis (Fig. 7.12). Squash preparations will often highlight the delicate hairlike (pilo [L.] hair) processes and granular bodies or protein droplets (Fig. 7.13). Rosenthal fibers are frequently seen in the compact areas (Fig. 7.14) and are diagnostically useful when present, although sometimes they are few in number (Fig. 7.15). Rosenthal fibers are a characteristic degenerative change found in pilocytic astrocytomas of all locations. They are not exclusively associated with pilocytic astrocytomas and may occasionally be found in other glial tumors including ganglioglioma and pleomorphic xanthoastrocytoma. They are also commonly found in association with chronic reactive gliosis, for example, in the regions adjacent to craniopharyngiomas, capillary hemangioblastomas, and syringomyelia cavities, within pineal cysts, and, in Alexander disease, a neurodegenerative disease of childhood characterized by absent myelination and numerous and often perivascular Rosenthal fibers. Rosenthal fibers are opaque, homogeneous, strongly eosinophilic, and corkscrew or beaded masses, and the vertebrate lens protein αβ-crystallin is a major component of them. In 1898, Rosenthal reviewed an intramedullary spinal cord tumor associated with a gliotic syringomyelia and described the fibers which would eventually become eponymously linked to him [47, 48].
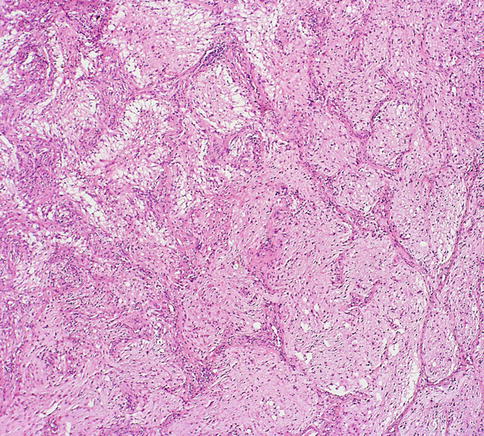
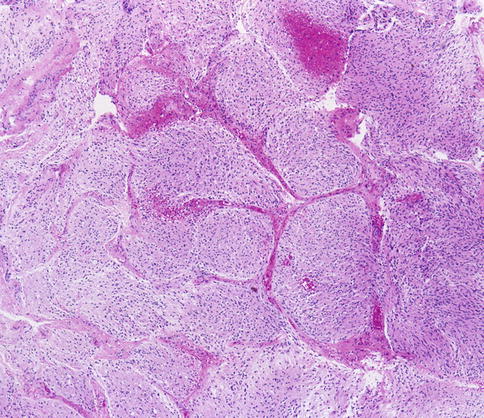
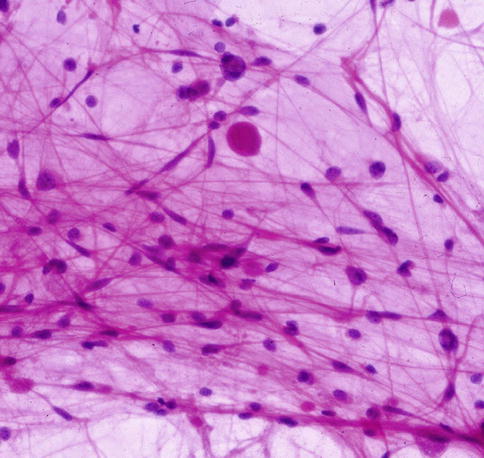
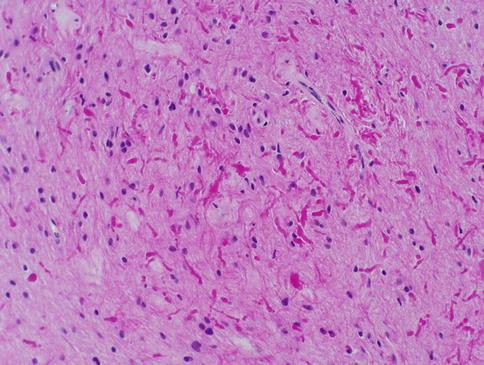
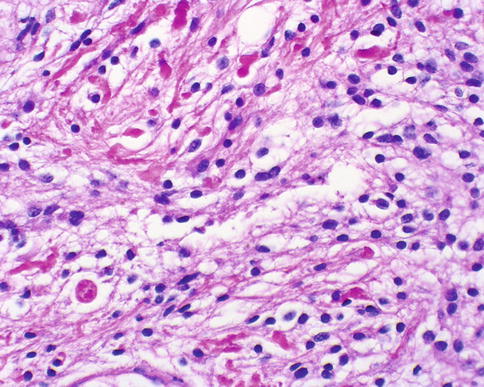
Fig. 7.11
Pilocytic astrocytoma of the optic nerve showing expansion of the septa (From Cummings [171], Figure 8.3, Page 158. With kind permission of Springer Science + Business Media)
Fig. 7.12
Low-magnification view showing expansion of the optic nerve septa by glioblastoma (From Cummings [171], Figure 8.12, Page 163. With kind permission of Springer Science + Business Media)
Fig. 7.13
Cytology squash preparation of an optic nerve pilocytic astrocytoma showing bipolar delicate hairlike processes and a round protein droplet. Hematoxylin and eosin staining is most useful in cytology preparations of brain tumors (From Cummings [171], Figure 8.4, Page 158. With kind permission of Springer Science + Business Media)
Fig. 7.14
Pilocytic astrocytoma of the optic nerve with numerous Rosenthal fibers. A case like this might be challenging to distinguish from reactive piloid gliosis (From Cummings [171], Figure 8.6, Page 159. With kind permission of Springer Science + Business Media)
Fig. 7.15
Pilocytic astrocytoma of the optic nerve showing bright pink waxy Rosenthal fibers. A round eosinophilic granular body is seen towards the bottom left (From Cummings [171], Figure 8.5, Page 159. With kind permission of Springer Science + Business Media)
Most optic nerve gliomas are pilocytic astrocytomas that resemble equivalent tumors in other sites of the central nervous system, but these tumors can be a diagnostic challenge for the surgical pathologist because of histological heterogeneity among and within tumors. Verhoeff described three histological patterns of optic nerve pilocytic astrocytomas including the coarsely reticulated, coarsely fibrillated, and finely reticulated types [49]. These patterns are recognized as variants of pilocytic astrocytomas, and although transition types and combinations of patterns can be found in any given tumor, usually one pattern predominates. The coarsely reticulated pattern is characterized by the classic biphasic pattern of coarse bipolar astrocytic cells either tightly compacted around blood vessels or loosely woven around microcystic spaces containing abundant mucin. Tumors with this histological pattern are commonly associated with Rosenthal fibers, eosinophilic granular bodies, and protein droplets. Blood vessels vary in number and may be hyalinized. The coarsely fibrillar pattern (also known as the spindle-cell type), consists of coarse neuroglial fibrils and spindle neuroglia cells generally arranged in fairly definite bundles. This pattern appears to describe the so-called adult variant of pilocytic astrocytoma as described by Rubinstein [50]. The finely reticulated pattern resembles an expansion of the indigenous neuroglia of the optic nerve, in which small round or ovoid nuclei are embedded within a delicate reticulated syncytium of neuroglial fibers. This pattern can be confused histologically with a well-differentiated infiltrative fibrillary astrocytoma (WHO grade II).
Immunohistochemical staining for glial fibrillary acidic protein [GFAP] highlights the tumor cells and their processes in optic nerve gliomas confirming a glial origin. The Ki-67 (Mib-1) proliferation-related labeling index of pilocytic astrocytomas of the optic nerve and other sites of the central nervous system is often low, less than 1 % [51, 52]. Some reports of optic nerve pilocytic astrocytomas, however, have documented elevated Mib-1 indices [52–57], which appears to agree with the natural history of many of these neoplasms that increase in size and then stabilize, especially in individuals younger than 20 years of age.
Differential Diagnosis
Potential pitfalls in pilocytic astrocytoma include piloid gliosis which can mimic and be difficult to distinguish from pilocytic astrocytoma. Optic nerve gliosis may include arachnoid gliomatosis [42] and diffuse hyperplastic gliosis and may be seen in patients with and without neurofibromatosis type 1 (NF1) [27, 58]. Diffuse leptomeningeal glioma may rarely involve the optic nerve sheath [59]. Optic nerve gliomas can be associated with meningothelial hyperplasia, and the pathologist must be aware of a superficial biopsy of the meninges to avoid making an incorrect diagnosis of meningioma [60]. On a tiny biopsy, the histological distinction of pilocytic astrocytoma from a diffuse or fibrillary (WHO grade II) astrocytoma might not be possible. Recent molecular advances have shown optic nerve gliomas with pilocytic features to harbor KIAA1549:BRAF fusions typical of pilocytic astrocytomas in other locations, resulting in mitogen-activated protein kinase (MAPK) pathway activation and signaling. In contrast to diffuse or infiltrating astrocytomas, IDH1 and IDH2 mutations are absent in pilocytic astrocytomas [61].
Some pilocytic astrocytomas exhibit anaplastic features including an increased number of mitoses, vascular proliferation, and necrosis with pseudopalisading [62–65]. Beware of misdiagnosing malignant glioma (glioblastoma WHO grade IV) in these cases. Glioblastomas are malignant astrocytomas characterized histologically by cytological pleomorphism, mitoses, microvascular proliferation (Fig. 7.16), and necrosis with or without pseudopalisading (Fig. 7.17). Attention to the clinical setting and radiographic appearance is recommended in these case.
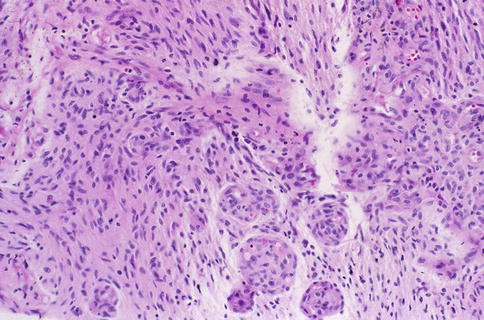
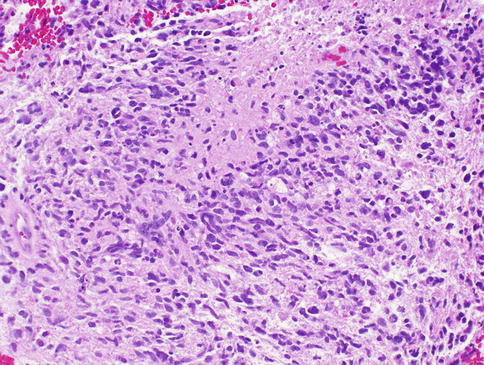
Fig. 7.16
Optic nerve glioblastoma showing microvascular proliferation (From Cummings [171], Figure 8.13, Page 164. With kind permission of Springer Science + Business Media)
Fig. 7.17
Optic nerve glioblastoma showing pseudopalisading necrosis (From Cummings [171], Figure 8.14, Page 164. With kind permission of Springer Science + Business Media)
Pilomyxoid astrocytoma (WHO grade II) is closely related to pilocytic astrocytoma, but these tumors are more aggressive and usually involve the hypothalamic/chiasmal region in young children. Rosenthal fibers and eosinophilic granular bodies are typically absent, and the tumor cells have perivascular orientations that resemble ependymal perivascular pseudorosettes (Fig. 7.18).
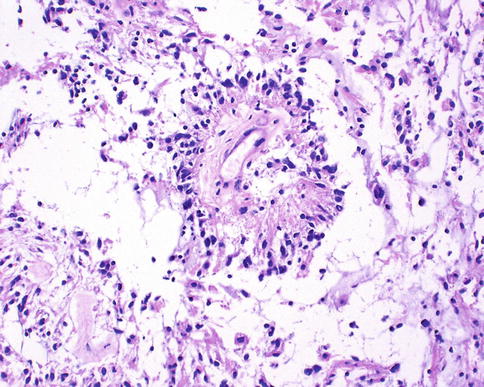
Fig. 7.18
Pilomyxoid astrocytoma characterized by perivascular tumor cell accumulations not to be mistaken for perivascular pseudorosettes of ependymoma (From Cummings [171], Figure 8.8, Page 161. With kind permission of Springer Science + Business Media)
Although oligodendrocytes are part of the normal cellular composition of the optic nerve, oligodendroglial neoplasms rarely have been reported to involve the optic nerve and chiasm [26, 28, 66, 67]. Gliomatosis cerebri is a rare diffuse WHO grade III glioma characterized by an extensive infiltration of neoplastic cells that do not form a defined mass lesion but can involve the supratentorial, infratentorial, and intraspinal compartments [68]. The tumor cells diffusely invade the brain parenchyma and frequently migrate along myelinated white matter fiber tracts including the optic pathway. Gliomatosis cerebri uncommonly presents with ophthalmological signs and symptoms including visual loss, diplopia, nystagmus, and homonymous hemianopia [69]. Few reports of gliomatosis cerebri have documented involvement of the optic nerves and chiasm [70–73].
Genetics
NF1 has a well-recognized association with tumors of the central nervous system, especially gliomas of the optic pathways [26, 27, 39, 74–82]. About 70 % of optic pathway gliomas are associated with NF1 [39], and these tumors have a generally more favorable outcome than non-NF1-associated gliomas, as most tumors grow slowly or not at all [27, 39, 58, 74, 79, 83, 84]. Approximately 20 % of optic nerve tumors affect both nerves in patients with NF1 [25, 85], yet extensive bilateral involvement of the visual tracts is rare [86]. Nonetheless, tumor enlargement and progressive visual impairment occurs occasionally in children with NF1 [39, 77, 84, 87] Although exceptional, malignant change has been reported in optic nerve gliomas in patients with NF1 [66, 88] or with a family history of NF1 [89]. Pilocytic astrocytomas of the optic pathway may show deletions and allelic losses involving the NF1 gene on the long arm of chromosome 17 and upregulation of NF1 gene transcripts, and it is thought that different mechanisms involving the NF1 tumor suppressor gene may play an important primary genetic event in the promotion or initiation of NF1-associated pilocytic astrocytomas [90–96]. NF1-associated pilocytics typically lack the genetic changes associated with aggressive fibrillary astrocytomas [97]. Although p53 protein accumulations can be detected by immunohistochemistry, mutations of the TP53 gene are rare [92, 98–101]. Molecular profiling using oligonucleotide microarray analysis has been able to demonstrate distinct gene expression patterns in pilocytic astrocytomas. Compared with glioblastomas, genes that encode cell motility were expressed at lower levels, while genes involved in suppressing migration were expressed at higher levels [102].
Prognosis and Predictive Factors
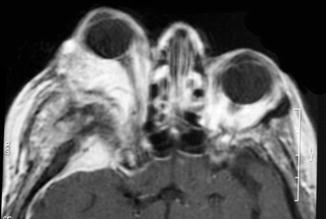
Pilocytic astrocytomas typically occur in children and young adults, and they are usually slow growing with a favorable prognosis. Despite incomplete surgical resection, some remain quiescent for extended periods of time and fail to recur, suggesting either arrest of tumor progression or deceleration in growth. Neuroimaging studies have documented spontaneous regression of large, clinically symptomatic optic pathway pilocytic astrocytomas [103–106]. Theories as to why these tumors spontaneously regress include individual tumor cell programmed death outpacing cellular proliferation, decreased vascular engorgement of the tumor, and resorption of the tumor-secreting mucosubstance [103, 107]. Some tumors, however, despite years of relative quiescence, retain the ability to grow and invade adjacent structures such as the optic chiasm and hypothalamus, resulting in considerable morbidity and mortality and a poorer prognosis than gliomas confined to a single optic nerve [24, 39, 46, 57, 74, 87, 108–112]. Others rarely exhibit malignant transformation and progression to anaplastic astrocytoma or glioblastoma [108]. Optic nerve glioblastomas typically occur in older adults, and patients present with unilateral or bilateral visual loss that often progresses to blindness. Glioblastomas are highly invasive and usually fatal within 1 year [31, 33]. Tumor may spread via the optic radiations to the occipital pole (Figs. 7.19 and 7.20).
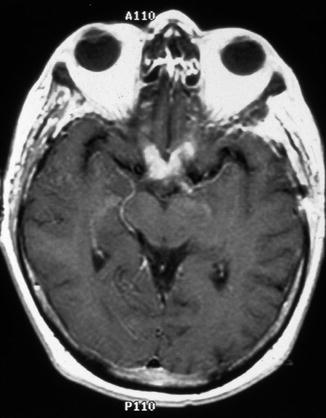
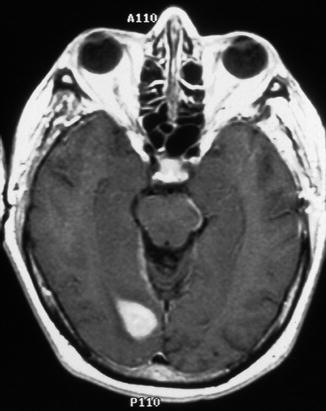
Fig. 7.19
Optic nerve glioblastoma. Contrast-enhanced T1-weighted axial magnetic resonance imaging (MRI) of the brain showing enhancement of the optic nerves and chiasm (From Cummings [171], Figure 8.9, Page 162. With kind permission of Springer Science + Business Media)
Fig. 7.20
Contrast-enhanced T1-weighted axial MRI of the brain showing extension of glioblastoma along the optic radiations to the visual cortex in the occipital pole (From Cummings [171], Figure 8.10, Page 162. With kind permission of Springer Science + Business Media)
7.7.3 Optic Nerve Meningiomas
Definition
Meningiomas are tumors thought to arise from meningothelial cells in the arachnoid mater of the optic nerve leptomeninges.
Epidemiology
Similar to meningiomas elsewhere in the CNS, those arising in the optic nerve sheath and orbit occur more often in females than in males. Conceivably, the increased incidence in females may possibly be related to the known presence of progesterone and estrogen receptors in meningiomas [113]. Similar to intracranial and intraspinal meningiomas, the majority of primary orbital meningiomas present in the fifth decade of life. In the experience of some investigators [114], about one-fourth of the cases of primary orbital meningiomas become symptomatic before 10 years of age, in sharp contrast to intracranial meningiomas, which are rarely apparent before the age of 20 [115]. However, this early age of onset has not confirmed in other studies [116].
Etiology and Localization
Meningiomas may arise anywhere along the optic nerve sheath (Fig. 7.4) (see also page 640), but the vast majority found within the orbit are direct extensions of intracranial meningiomas from along the wing of the sphenoid bone (sphenoidal ridge meningioma) [117, 118], the tuberculum sellae (suprasellar meningioma), or above and adjacent to the cribriform plate (olfactory groove meningioma) [119]. Sphenoorbital meningiomas (SOMs) involve the sphenoid wing, orbit, and cavernous sinus. They typically are associated with exuberant hyperostosis of the sphenoidal wing and direct involvement of the orbit (Fig. 7.21). The hyperostosis is due to direct tumor invasion of the bone [120]. SOMs represent up to 9 % of all intracranial meningiomas, and they are associated with a high rate of recurrence, up to 35–50 %. Clinically, patients with SOMs typically present with headaches, focal retro-orbital pain, proptosis, visual field defects, and ocular paresis. Ectopic orbital meningiomas may reside outside the muscle cone without an attachment to the optic nerve [114, 121] or rarely external to the periosteal coverings of the orbital walls. Orbital meningiomas are seldom bilateral [122]. Other meningiomas of ophthalmological importance sometimes arise beneath the conjunctiva or caruncle [123], within the optic canal [124], paranasal sinuses, and the skin around the eye [125].