Ophthalmic Manifestations of Defects in Metabolism
J. Bronwyn Bateman
This chapter describes most of the multisystem disorders that have a metabolic basis and reported ocular manifestations. Although most are autosomal recessive or X-linked recessive, some metabolic disorders are autosomal dominant or X-linked dominant. The terms disease, disorder, and syndrome are used interchangeably, based on historical precedent. The human genome project has provided the infrastructure for the identification of all human genes and genetic diseases. Thus, the definition of a “metabolic” disease has broadened and now encompasses all gene defects. For the purposes of this chapter, only those diseases that are Mendelian or single gene, associated with multisystem disease and are “metabolic” in the more traditional definition are included. Organization of the chapter is by biochemical pathways and systems.
Most genetic disorders are “germinal” and the DNA defect is found in all cells of the organism (individual), including the reproductive cells (ova and spermatozoa in humans); others are “somatic.” Somatic genetic defects are found in a group of cells such as occurs in the nonhereditary form retinoblastoma in which only a portion of the cells, usually the tumor cells, contain the defect. Mosaicism refers to genetic differences in a percentage of cells in the organism (e.g., only white cells in the blood) or in a percentage of all cells. This chapter will include the traditional metabolic defects causing multisystem and ocular manifestations inherited in a Mendelian pattern (i.e., single gene defects). This group encompasses diseases that are expressed in many cell types and tissues, caused by single gene defects, and usually inherited in an autosomal recessive or X-linked recessive Mendelian pattern. Some of the diseases in this group have a similar phenotype or expression with more than one gene causing the phenotype. As an example, the phenotype of retinitis pigmentosa can be caused by multiple gene defects. Conversely, different mutations of a single gene, alleles, can produce different phenotypes, and these phenotypes can have different names. Further complicating nomenclature, the identical mutation of the same gene can produce different phenotypes, caused by the effect of other genes and/or environmental factors. The genetic nomenclature is now standardized and accessible through the Online Mendelian Inheritance of Man (OMIM). This numerical system is authored by Victor McKusick of Johns Hopkins University and his colleagues; it was initiated in a 1966 publication, the Mendelian Inheritance in Man. This Internet database is authored by McKusick and numerous colleagues, maintained by the National Center for Biotechnology Information (NCBI) and funded by a contract from the National Library of Medicine and the National Human Genome Research Institute in the United States. The OMIM database is organized numerically and includes a Gene Map that presents cytogenetic map location of disease genes and other expressed genes and a Morbid Map that presents the cytogenetic map location of disease genes. Each OMIM entry is given a unique six-digit number; the first digit indicates the mode of inheritance of the gene involved in the disease:
1—– (100000-) Autosomal loci or phenotypes (entries created before May 15, 1994)
2—– (200000-) Autosomal loci or phenotypes (entries created before May 15, 1994)
3—– (300000-) X-linked loci or phenotypes
4—– (400000-) Y-linked loci or phenotypes
5—– (500000-) Mitochondrial loci or phenotypes
6—– (600000-) Autosomal loci or phenotypes (entries created after May 15, 1994).
The symbols preceding the number are defined as follows:
An asterisk (*) before an entry number indicates a gene of known sequence.
A number (#) symbol before an entry number indicates that it is a descriptive entry, usually of a phenotype, and may not represent a unique locus. The reason for the use of the #-sign is given in the first paragraph of the entry. Discussion of any gene(s) related to the phenotype resides in another entry(ies) as described in the first paragraph.
A plus (+) sign before an entry number indicates that the entry contains the description of a gene of known sequence and a phenotype.
A percent (%) sign before an entry number indicates that the entry describes a confirmed Mendelian phenotype or phenotypic locus for which the underlying molecular basis is not known.
The database can be accessed at http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/. When feasible, the symbol and number of the disease, gene, and/or phenotype is included in this chapter. In the text, the phenotypes reviewed are diseases.
Generalizations are hazardous in genetics as the field increases in complexity as genes and mechanisms are identified at an exponential rate. However, most autosomal recessive diseases are caused by mutations of proteins that are enzymes, have developmental cellular functions, or regulate the energy pathway; these diseases may be caused by a deficiency of the protein or a “dominant negative” effect (see below). Most autosomal dominant diseases are caused by mutations of proteins that have a structural function; however, such diseases may occur as a result of splicing errors, transcription, or cell growth regulation functions. The mutant structural protein can be disruptive to tertiary structure and have a dominant negative effect. An example of an autosomal dominant negative effect is Marfan syndrome in which the structural protein fibrillin, the protein that accounts for most if not all cases of Marfan syndrome, is defective as a result of a DNA sequence defect; although reduction of the amount of normal fibrillin may be a factor in the disease, a dominant-negative effect is likely with the mutant fibrillin protein disrupting the function of the normal, wild-type protein. A relatively new basis for autosomal dominant disorders is expansion of nucleotide repeats in untranslated regions of genes (introns). The basis for myotonic dystrophy and several other disorders is the expansion of normal numbers of DNA repeats (such as cytosine, thymine, guanine [CTG]) from one generation to another. This expansion can increase from one generation to the next and cause the disease to become more severe in subsequent generations (a rare example of the concept of anticipation defined as an increased severity of disease from one generation to the next). Conversely, the phosphodiesterases are retinal enzymes in the visual transduction cascade and mutations cause an autosomal recessive disease; the reduction in the amount of the protein causes the disease. However, there are many examples of exceptions. The photoreceptor peripherin gene, producing a membrane-associated glycoprotein, causes disease if one or both alleles (an allele is the DNA encoding for the gene on a single chromosome and any form (normal (wild-type), common variation such as blood type or disease-causing change) is an allele) has an abnormal sequence; thus, retinal disease, as a result of peripherin gene mutations, occurs in autosomal dominant and recessive patterns. Proteins in any subcellular location may be associated with metabolic disease including the lysosome, microsome, endoplasmic reticulum, Golgi apparatus, cell surface, and even the nucleus. Enzymatic diseases are more likely to occur if there is less than 50% activity of the protein, and abnormal regulatory proteins may cause disease by altering embryogenesis or cellular regulatory mechanisms. The abnormal and less functional enzymatic proteins that occur in the heterozygote or carrier usually have approximately 50% activity; the normal allele is translated into a normal protein and the mutated allele is abnormal and may have some enzymatic activity. Thus, in most cases, the carrier (heterozygote) of a mutation in a gene encoding an enzyme is protected from the disease. Males have one X chromosome and have diseases termed X-linked recessive if a gene on the X chromosome is mutated (hemizygote); female carriers may be unaffected or develop the manifestations later in life. X-linked dominant diseases occur in both males (hemizygote) and females (heterozygote) if one X chromosome allele is mutated; frequently, fetal wastage (intrauterine demise) of males occurs. A null mutation is one in which no protein product is made. Geographically, culturally, or religiously isolated populations may have a higher incidence of a given disease and a limited number of mutations; this is termed the “founder” effect and is due to DNA from a common ancestor.
Additionally, some genes may be predisposed or, alternatively, relatively resistant to mutations. The DNA sequence, size of the gene, homology of the DNA sequence of the gene to other genes, position on a chromosome, potential for morbidity to the cell or embryo, and other unknown factors influence the frequency of mutations and the prevalence of the gene defect and disease in a population. If a disease occurs as a result of many different mutations of the same gene, it is termed “private,” indicating that it is particular to an individual or family. Some mutations provide evolutionary advantages, such as sickle cell disease that is a genotype protective of malaria in the heterozygous state (one normal allele and one abnormal allele).
Further complicating Mendelian patterns of inheritance, phenotypic features of different mutations of the same gene may vary considerably. Mutations of the fibrillin gene can cause tall stature, long, thin digits, joint laxity, and dislocated lenses, typical features of Marfan syndrome; other mutations may cause short stature, brachydactyly, and dislocated lenses, typical features of Weill-Marchesani syndrome.
Eponyms are now used less frequently and diseases are categorized by the gene defect; increasingly, the specific mutation is identified and the prognosis may be estimated if genotype-phenotype correlations have been established. Using easily accessible cells such as white blood cells, either mutational analysis of likely (candidate) genes or biochemical confirmation are necessary for diagnosis; the conjunctiva biopsy is ideal for identifying a category among the inborn errors of metabolism.
Clinical features and inheritance patterns are a function of the specific mutation, posttranslational differences, tissue expression, and other genetic and environmental influences. An example of environmental influences on genetic disease is galactosemia; if the disease is diagnosed early in life, the exclusion of galactose from the diet reduces the risk for mental retardation and cataract formation.
Although a diagnosis can be considered clinically for many of the metabolic diseases in this chapter, the specific diagnosis is usually made on the basis of DNA or protein studies. A combination of ocular examination, conjunctival biopsy, and/or electroretinogram may be very useful to the geneticist. As mental retardation is commonly associated with strabismus and cortical visual inattentiveness, those metabolic disorders in which mental retardation is associated only with strabismus or cortical visual have not been included in this chapter. Chromosomal rearrangements and disorders that are unlikely to be genetic, such as Sturge-Weber syndrome, have been specifically excluded. For some metabolic diseases in which the manifestations are severe and can be prevented or the clinical course modified, newborn screening has been legislated at the state level. Although there has been considerable controversy as to which tests should be performed in a newborn, the required tests are increasing in number. Historically, the specific tests required of every newborn are under the jurisdiction of individual states (specific tests for each state in the United States are available at http://www.genes-r-us.uthscsa.edu). Additionally, certain tests are recommended for specific ethnic groups or in specified geographic regions in the US and the world.
For many metabolic diseases, treatment of the patient and genetic counseling for the family for monitoring of future pregnancies are feasible. Prenatal diagnosis has been performed in many diseases and should be feasible in all in which the gene and the mutation can be determined by DNA sequencing.
For diseases in which the gene is known and for families in which the specific gene defect is known, preimplantation genetic diagnosis is feasible to ensure a newborn will be born without the disease. These diseases should be amenable to preimplantation testing in which one or a few cells from in vitro (test tube) fertilized blastosphere can be tested for the defect, and those blastospheres without the gene defect can be implanted in the uterus for gestation.
Gangliosidoses and Lipid Storage Disorders
The lysosomal storage diseases are a class of metabolic disorders resulting from deficiencies of lysosomal enzymes. Metabolites of complex lipid accumulate within lysosomes, located in the cytoplasm. Previously fatal, some forms are now treated with bone marrow transplant.
Sphingolipidoses
The sphingolipidoses are a group of neuronal lipid storage disorders clinically characterized by a progressive degeneration of the central and peripheral nervous system and are due to deficiencies of lysosomal enzymes with accumulation of normal lipid or glycolipid; in most cases the defect is in a lysosomal hydrolase. Lipids are critical in all biological membranes and for normal cellular function. Sphingolipids are complex molecules with a ceramide sphingogosine and attached fatty acids, at the structural core; in most, sugar molecules also are attached. The sphingolipids include sulfatides, sphingomyelins, and gangliosides. The shared pathway of the sphingolipidoses involves reduced activity of degradative enzymes or activator proteins for these enzymes, resulting in accumulation of lipid compounds. These degradative enzymes act sequentially and, if activity of one is reduced or absent, the sphingolipid degradation is interrupted. The stepwise enzymatic degradation of these molecules results in accumulation, at least partly within lysosomes, of the accumulated products.1 Sphingolipids are expressed in neural and nonneural tissues. The disease associated with the degradative processes is a function of the tissue distribution of the particular sphingolipids. Those disorders that affect the neural tissues are usually divided by the neural tissue involved in the early stages of disease and categorized into the leukodystrophies (white matter), myelin-containing cells, and the neuronal (gray matter) processes. The sphingolipids found primarily in white matter are galactosylceramide and sulfatide; a common clinical feature is progressive loss of motor milestones. The clinical features of gray matter disorders include seizures, progressive mental retardation, and visual impairment.
The diseases included in the sphingolipidoses include the GM1 and GM2 gangliosidoses, the Niemann-Pick disorders, Gaucher disease, Farber disease, metachromatic leukodystrophy, and Krabbe disease. Most affected individuals are normal at birth and the age of onset for signs or symptoms is related to the specific mutation or combination (compound heterozygote for autosomal recessive disorders with two different alleles). All are autosomal recessive except Fabry disease, which is X-linked recessive.
Gangliosidoses and Related Lipid Storage Diseases
Gangliosides have an N-acetylneuraminic acid (NANA) linked to the hexose units and vary by the length of the sugar and the number of NANA groups; more than 15 have been identified and four constitute over 80% of the total in humans.2 Metabolism of gangliosides involves the removal of the terminal galactose to convert GM1 ganglioside to GM2 ganglioside. GM2 ganglioside is then hydrolyzed to GM3 ganglioside by the removal of N-acetylgalactosamine. Ten ganglioside storage diseases are now recognized. Seven of the disorders involve storage of GM2 ganglioside, and three involve the accumulation of GM1 ganglioside.
GM1-gangliosidosis (OMIM +230500)/Morquio, type B (mucopolysaccharidosis type IVB) (OMIM #253010)/(gangliosidosis, generalized GM1, type II; OMIM #230600)/(gangliosidosis, generalized GM1, type III, or adult type; OMIM #230650)
The accumulation of the gangliosides GM1 and asialo GM1 is caused by a deficiency of GM1 β-galactosidase, the gene (GLB1) for which is on chromosome 3p21.33. The diseases, GM1 gangliosidosis3,4 and Morquio syndrome type B,4,5 are inherited as autosomal recessive traits, result from mutations of this gene, and cause an inability to cleave the terminal galactose from several molecules including GM1 ganglioside, galactose containing oligosaccharides, and keratin sulfate. The absence of β-galactosidase isoenzymes A, B, and C leads to the storage of a variety of macromolecules with a terminal β-galactosyl residue, primarily GMI ganglioside and complex carbohydrates. The accumulation of complex carbohydrates is the basis for the visceral and skeletal manifestations. Because β-galactosidase can be a monomer or a dimer and is in a multiprotein complex with neuraminidase and a protective protein, manifestations vary with differences in synthesis, posttranslational modification, and degradation,6,7 and are a result of different mutations.4,8,9,10,11,12
The diseases, generalized gangliosidoses or Hurler variant, occur in an infantile, late infantile, and adult form, depending on the age of onset and severity. The infantile form has a relatively early onset in infancy with early death; manifestations of edema and hypotonia may be evident at birth. The progressive manifestations include psychomotor retardation, hepatosplenomegaly, and bony abnormalities. The infantile form is characteristic of a storage disease with coarse features, frontal bossing, macrocephaly, and a protuberant abdomen due to enlargement of visceral organs. The juvenile and adult forms are less common with onsets from ages 6 to 12 months and from ages 3 to 50 years, respectively.13 Neurologic symptoms are the initial manifestation with progression more variable in the adult form; affected individuals with the juvenile form usually have an onset in childhood. The Morquio B form has similar clinical features of the mucopolysaccharidosis Morquio A (different gene; see separate entry) with dwarfism and bone dysplasia but a later onset and milder phenotype; intelligence is normal.14,15
Visual inattentiveness is an early sign of the infantile form of generalized gangliosidosis or Hurler variant. The ocular features include hypertelorism, ptosis, strabismus, lid edema, corneal clouding, a “cherry-red spot” in the retina, tortuous retina vessels, retinal hemorrhage, and optic atrophy.16,17 Nystagmus may develop. The ocular features of the juvenile and adult forms are less prominent.18 The ocular features of the Morquio B include corneal clouding.14,15
GM2-gangliosidoses (OMIM #272800) (OMIM +27275) Tay-Sachs disease; Sandoff disease (OMIM #26880) Tay-Sachs disease, AB variant (OMIM +272750)
The GM2 gangliosidoses are a group of autosomal recessive diseases caused by the deficiency of β-hexosaminidase [composed of an alpha20,21 (HEXA) (OMIM *606869) and a beta (OMIM *606873) subunit] or the GM2 activator (OMIM +27275) with accumulation of GM2 gangliosides. The three genes encoding these proteins, β-hexosaminidase, α-hexosaminidase, and the GM2 activator (GM2A) are on chromosomes 15q23-24, 5q13, and 5q31.3-33.1, respectively. These enzymes are the degradative pathway for GM2 ganglioside and a deficiency causes failure to hydrolyze the terminal amino sugar. Hexosaminidase A is a thermolabile protein composed of two alpha and two beta subunits; the hexosaminidase B is thermostable and contains four beta subunits. Both forms hydrolyze N-acetylhexosamine from glycoproteins, glycolipids, glycosaminoglycans, and oligosaccharides. The Hex A form hydrolyzes only the GM2 ganglioside; the Hex B form metabolizes the GM2 ganglioside and neutral sphingolipid globoside. Deficiency of the α subunit results in the Hex A only (Tay-Sachs) and reduced function of the β subunit causes deficiency in both enzymes (Sandoff).22 Deficiency of each of the three proteins causes accumulations of GM2 ganglioside. The GM2 activator deficiency is the rarest form and is a variant clinically.23
The clinical features are caused by the accumulation of GM2 in brain, liver, and spleen tissue with the asialo and lyso forms increased in the brain. The lyso GM2 is cytotoxic. Although identification of the carrier state and prenatal diagnosis are feasible, affected individuals have few treatment options. Bone marrow transplant has been attempted with limited success.24
β-hexosaminidase, α-subunit deficiency (Tay-Sachs) (OMIM #272800)
Named after a British ophthalmologist, Tay,25 this autosomal recessive disease is characterized by an acute onset, usually between the fifth or sixth month of age with progressive neurological deterioration and death, generally, by 5 years of age. The diagnosis of an inborn error of metabolism is commonly made on the basis of an ophthalmologic exam for visual inattentiveness, an early sign, with a “cherry-red spot” in the maculae. The cherry-red spot, evident on ophthalmoscopy, occurs because the fovea is devoid of ganglion cells. The cherry-red spot is caused by accumulation of GM2 in retinal ganglion cells; normal foveal color appears red in contrast to the surrounding pale tissue. Macrocephaly may occur as a result of gliosis (as opposed to accumulation of the GM2 ganglioside).
The juvenile and chronic forms are caused by different mutations. Because of the founder effect, the disease is more common in the Ashkenazic Jewish and French-Canadians populations. Although many mutations of the early onset form have been described, 80% of Ashkenazic Jews have the same mutation; three mutations account for 98% in Ashkenazic Jewish patients.26,27,28
Clinically, the onset of the disease occurs early in life in the classic, infantile form and death is usually in infancy. Diagnosis may not be made until later in childhood when the initial signs of either cerebellar or anterior motor neuron disease are evident. Affected individuals may also develop psychotic episodes. The basis is residual activity in at least one allele.29
The ocular features include the cherry-red spot in the maculae. The cherry-red spot may disappear over time as the ganglion cells die and become atrophic, resulting in a disappearance of the red appearance in the fovea (Fig. 1).30 Gangliosides accumulate in the retina and corneal endothelium.31 Abnormalities of the oculomotor system and saccadic velocities may be evident in the late onset form.32,33
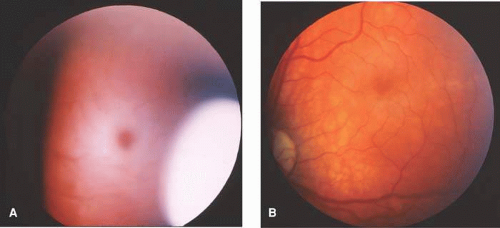 Fig. 1. Tay-Sachs disease. a: Cherry-red spot (normal fovea with creamy appearance of macular region due to accumulation of GM2 gangliosides in the ganglion cells). b: Normal appearing fundus later in the disease of the same patient due to atrophy of retinal ganglion cell layer. (Courtesy of Jane D. Kivlin MD) |
α– and β-hexosaminidase, β-subunit deficiency (Sandhoff disease) (OMIM #268800)
Sandhoff disease is an autosomal recessive disease caused by deficiency of both α– and β-hexosaminidases; the gene (HEXB) (OMIM *606873) encodes β-subunit protein. There is no ethnic predisposition. Although not as well studied as Tay-Sachs, many of the infantile forms have a common deletion.34 Considerable phenotypic variability has been demonstrated in patients with the identical mutation.35
The clinical features are similar to those of Tay-Sachs disease with loss of developmental milestones early in infancy, and progressive neurologic degeneration leading to death. Forms with a later onset have been described and are very rare.
GM2 gangliosidosis—AB variant (hexosaminidase activator deficiency; GM2 gangliosidosis, type AB) (OMIM +272750)
Deficiency of the activator protein, the transporter of GM2 from the lysosomal membrane to the enzyme, is inherited as an autosomal recessive trait.38 The protein binds to GM2, removes it from the membrane, and solubilizes it as an activator/lipid complex. The gene for this activator protein is on chromosome 5q31.3-q33.1. Rarer than the other two forms, several mutations have been described.23,39,40
This rare variant has clinical features similar to Tay-Sachs and Sandhoff diseases.38
The ocular features described in Tay-Sachs and Sandhoff diseases, the cherry-red spot in the maculae, have been reported.41
Sphingomyelin lipidoses (Niemann-Pick type A) (OMIM #257200) (Niemann-Pick type B) (OMIM #607616)
Sphingomyelins accumulate in the sphingomyelin lipidoses; both forms, A and B, are inherited in an autosomal recessive pattern and caused by mutations of the gene encoding the enzyme sphingomyelinase (SMPD1) (OMIM *607608) on chromosome 11p15.1-15.4.42 Type A is primarily a Jewish disease and over 90% of the mutations in this population are point mutations or a single nucleotide deletion (R496L and L302P and fsP330).43,44,45,46,47 A three-base pair in frame deletion (delR608) is associated with type B (homozygous or heterozygous with the R496L).44,48,49 Type B disease occurs in different populations and multiple mutations have been described.47,50,51 Small deletions that truncate the protein or missense mutations that make the protein noncatalytic cause type A disease.47 Although a molecular diagnosis is ideal, conjunctival biopsy is useful as the pathologic ocular features are characteristic.52,53,54 Preimplantation genetic diagnosis is feasible.55
The deficit of lysosomal acid sphingomyelinase results in intracellular accumulation of sphingomyelin. Pathologically, there is accumulation of “foam cells” throughout the reticuloendothelial system.
Clinically, the two forms are differentiated by the age of onset (type A in early infancy and type B in early childhood) and the neurological symptoms (type A with progressive psychomotor deterioration and type B without neurological involvement). Both forms are associated with hepatosplenomegaly.
In the type A form, the patient develops spasticity, rigidity, and death usually occurs before the age of 4 years. Individuals with type B develop interstitial pulmonary disease, cirrhosis, hyperlipidemia, and fractures.47,56 The clinical features are related to the specific mutation.
Glucosylceramide lipidoses (Gaucher disease) type I (OMIM #230800); type II (OMIM #230900); type III (OMIM #231000)
Gaucher disease (GD) is characterized by the accumulation of glucosylceramide. The glucosylceramide lipidoses are inherited in an autosomal recessive pattern. The deficient enzyme is glucosylceramide β-glucosidase (acid β-glucosidase or glucocerebrosidase) and the gene, glucosidase beta acid (GBA) (OMIM *606463) is on chromosome 1q21. The protein catalyzes the hydrolysis of glucosylceramide to ceramide and glucose, and requires saposin C (SAP-C), an activator protein. Gaucher disease is due to mutations of the GBA gene and has been divided into three subtypes. The adult or chronic form, type I (OMIM #230800) accounts for approximately 85% of cases. The more severe infantile form, type II (OMIM #230900) (10% of cases) is neuropathic and the later onset (nonjuvenile) form, type III (OMIM#231000) is primarily from Sweden (Norrbottnian).62,63,64 The more common adult form is more common in the Ashkenazic Jewish population; at least seven mutations account for over 90% in this population and are probably due to founder effects.65,66,67 All three forms are caused by mutations of GBA, and the clinical manifestations differ as a result of the specific mutation68,69,70,71,72,73,74,75,76 and other factors. There is limited correlation between the genotype and phenotype69 and significant intrafamilial variability has been described. Hundreds of mutations have been described and some are believed to be caused by a process of gene conversion in which the normal gene exchanges DNA with a nearby nonfunctional duplication of the gene with resultant abnormal sequences in the functional gene.69,77 Although accumulation of the lipid glucosylceramide is the presumed basis of the disease, it accounts for less than 2% of the increased tissue mass and an inflammatory component is likely.78,79 Rarely, a mutation of the prosaposin gene (PSAP) (OMIM #176801) results in a deficiency of saposin C80,81 or D82 and the clinical features of Gaucher disease (see entry for sulfatide lipidoses).
Enzyme replacement therapy is commercially available and effective for the nonneurologic manifestations; unfortunately, the enzyme does not cross the blood-brain barrier and the neurologic features are not ameliorated.83,84,85 Other modes of therapy include reduction of substrate, chemical chaperones to stabilize the enzyme, and gene therapy.84,86 Carrier status (heterozygote) appears to be a risk factor for the development of Parkinson disease.87
In the more common adult form of Gaucher disease (type I), symptoms usually begin in childhood with growth failure, progressive splenomegaly, and pancytopenia; some affected individuals are asymptomatic. Progression is variable and central nervous system (CNS) complications are not a primary feature88 but may develop as a result of bony and vascular effects.89 The infantile form (type II) has an onset in infancy, similar to Niemann-Pick disease with hepatosplenomegaly, and the disease progresses rapidly; fetal hydrops may be evident during the pregnancy. Major signs and symptoms include failure to thrive, loss of neurologic milestones, and hepatosplenomegaly; recurrent pulmonary infections are evident. Untreated, death usually occurs by age 2 years. In the juvenile form (type III), splenomegaly may develop early, but loss of neurological milestones occurs later in childhood. Death usually occurs in childhood.62
Early reports of a cherry-red spot antedate the biochemical diagnoses.90 The documented ocular feature of all forms include oculomotor apraxia.91 Horizontal supranuclear gaze palsy may occur in types II and III.92,93 Vitreous opacities94 and “white spots”90,95 in and on the retina have been described in type I that consist of swollen histiocytes (Gaucher cells). Chronic uveitis96 and a progressive retinal degeneration97 have been described.
Ceramide lipidoses (lipogranulomatosis; Farber disease) (OMIM +22800)
Ceramide lipidosis is an autosomal recessive disease in which the enzyme acid ceramidase is deficient and ceramide accumulates. The gene has been mapped to chromosome 8p22-p21.3 and several mutations have been described.98,99
Clinically, fetal hydrops may be evident during gestation.100 Early in life, a weak, hoarse cry; lymphadenopathy; joint swelling; and multiple subcutaneous nodules are evident. Hypotonia and progressive psychomotor retardation ensue, and death usually occurs relatively early in life. Milder variants may occur.101,102
Trihexosylceramide lipidoses (Fabry disease) (OMIM +301500)
Trihexosylceramide lipidoses is an X-linked recessive disease caused by deficiency of α-galactosidase A105 and the accumulation of trihexosylceramide106 and digalactosylceramide. Many mutations of the gene (GLA), located on chromosome Xq22.1, have been reported.107,108,109,110,111,112 Although enzymatic assay in leukocytes and cultured skin fibroblasts will identify most affected individuals, molecular analyses are more reliable. Enzyme replacement therapies are useful.113,114
Clinically, lipid deposits in the vascular endothelium are the basis of renal, cardiac, and cerebral diseases, the most serious manifestations; accumulations in lung, liver, and kidney tissues also occur.115,116 In 1898, the skin lesions were described by two authors, Anderson117 and Fabry.118 Affected males develop pain in the extremities (acroparesthesia) and fever, precipitated by exercise, stress, or sun exposure; these symptoms are attributed to a peripheral neuropathy. Characteristic angiokeratoma corporis diffusum, cutaneous vascular lesions, occur in the periumbilical region, lower back buttocks, hips, and thighs and represent lipid infiltration of vascular endothelium. In mildly affected patients with preserved renal function, the nerve damage involves small myelinated and unmyelinated fibers.119 The renal, cardiac, and cerebrovascular manifestations—renal failure, cerebral vascular accidents,120 and cardiac dysfunction—cause premature death in both the hemizygotes (affected males) and heterozygotes (affected carrier females).114,121,122 There is a correlation of mutation and residual enzymatic activity with the renal failure.121
Nearly all hemizygous (affected) males and more than 70% of heterozygous (carrier) females exhibit ocular manifestations.116 Corneal manifestations include a generalized haze and corneal verticillata, a whorllike opacification of the corneal epithelium that is similar to the sequelae of systemic amiodarone treatment for cardiac disease (Fig. 2),123,124 with haze being the most frequent.125 Nearly half of the patients have radiating posterior subcapsular opacities in a starlike pattern that do not reduce vision (Fig. 3); anterior capsular cataracts are evident in approximately one third and may cause visual impairment.124,125 Vascular abnormalities of the conjunctiva and retinal vessels are common. Retinal vein occlusion has been reported in a heterozygote female126 and central retinal artery occlusion in affected males and heterozygote females.124,127,128 Bilateral Horner syndrome129 and internuclear ophthalmoplegia130 has been described in the hemizygous (affected) males. Choroidal ischemia has been reported.131 Anterior ischemic optic neuropathy has been reported in a heterozygote female.127
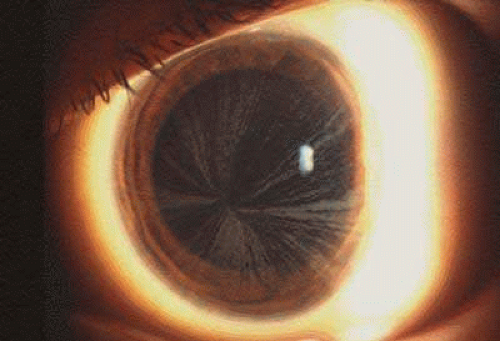 Fig. 2. Fabry disease. Corneal manifestations with visible whorl-like pattern of epithelial growth in an affected male. (Courtesy of Irene H. Maumenee MD) |
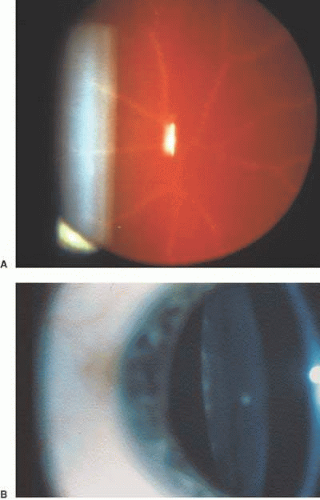 Fig. 3. Fabry disease. Star-like cataract at level of posterior capsule with indirect illumination. The vision is not affected by the lens opacity. (Courtesy of Irene H. Maumenee MD) |
Sulfatide lipidoses (metachromatic leukodystrophy) (OMIM #250100)
The sulfatide lipidoses (metachromatic leukodystrophy) are a group of autosomal recessive diseases in which sulfatide sulfatase (arylsulfatase A; ARSA) (OMIM #607574) or the sphingolipid activator protein (prosaposin gene) (PSAP) (OMIM +176801) is deficient. Saposin B, a proteolytically cleaved product of the prosaposin gene, is necessary in the hydrolysis of sulfatide by arylsulfatase A. The arylsulfatase A (ARSA) gene has been mapped to 22q13.31-qter and prosaposin to 10q22.1. Both proteins are required for the degradation of galactosyl-3-sulfate ceramide, a myelin membrane lipid. Galactosyl-3-sulfate ceramide and other galactosphingosulfatides accumulate in many tissues including brain. There is a progressive demyelination. Some mutations are associated with residual enzymatic activity,132 and the term pseudodeficiency is used for those alleles.133,134 In this disease, many mutations have been identified, and phenotype-genotype correlation has been established.133,135 The juvenile form is caused by pseudodeficiency mutations of the sulfatide sulfatase or mutations of PSAP.136 One polymorphism is more common in the African-American population but does not cause the phenotype of metachromatic leukodystrophy.137
In the most common form, late infantile, symptoms develop in the second year of life and death usually occurs before 5 years. Clinical features include progressive loss of both motor and CNS milestones with ataxia and hypotonia; spasticity develops and some individuals have seizures. The cerebrospinal fluid contains elevated protein. The juvenile form has an onset between 4 and 8 years of age with death in the second decade; initial symptoms include ataxia, cognitive impairment, personality changes, and seizures. Affected individuals may survive into early adulthood.132 The adult form has an onset in the late teenage years, usually with a psychiatric disorder; this form may be caused by heterozygotes with the pseudodeficiency allele and a true deficiency allele.134
The ocular features are limited. Ultrastructural study of the eye in a patient with the late infantile form showed a demyelinating process in the optic nerve, including the optic nerve head and intraneuronal storage of characteristic lysosomal residual bodies in ganglion cell perikarya of the retina.138 A patient with a variant form had cherry-red spot in the maculae.139
Multiple sulfatase deficiency (OMIM #272200)
Multiple sulfatase deficiency (MSD) is an autosomal recessive lysosomal disorder caused by mutations of the sulfatase modifying factor 1 (SUMF1) (OMIM #607939). The gene encodes the FGly-generating enzyme (FGE) that converts a highly conserved cysteine within the sulfatase catalytic domain into Cα-formylglycine140,141,142; the gene is conserved from pro- to eukaryote143 cells and mutations in humans cause multiple sulfatase deficiency.141,144 Biochemically, the activity of all known sulfatases is reduced.145 Sulfatases are a family of enzymes on multiple chromosomes that catalyze the hydrolysis of ester sulfates (including glycosaminoglycans, sulfolipids, and steroid sulfates); these proteins are located in different regions of cells.146,147,148 Cα-formylglycine (FGly), the catalytic residue in the active site of sulfatases, is posttranslationally generated from a cysteine in the endoplasmic reticulum.140 Several human diseases are caused by mutations of individual sulfatase genes: five mucopolysaccharidoses, metachromatic leukodystrophy, X-lined ichthyosis, and X-linked recessive chondrodysplasia punctata. Diagnosis may be made by enzyme assays or more accurately by sequence analysis of the gene. Several mutations have been reported.149
Clinically, the phenotype includes the features of all disorders caused by each of the single sulfatase deficiencies. The deficient enzymes include arylsulfatase A (ARSA) on chromosome 22q13 (metachromatic leukodystrophy), arylsulfatase B (ARSB) on chromosome 5q13-14) (Maroteaux-Lamy; MPSVI), arylsulfatase C (ARSC/STS) on chromosome Xp22.3 (X-linked ichthyosis), arylsulfatase D (ARSD) on chromosome Xp22.3 (isolated disease unknown), arylsulfatase E (ARSE) on chromosome 22.3 (chondrodysplasia punctata), arylsulfatase F (ARSF) on chromosome Xp22.3 (isolated disease unknown), iduronate S (IDS) on Xq27-28 (Hunter; MPSII), sulfamidase (HS) on chromosome 17q25.3 (Sanfilippo A; MPS IIIA), glucosamine 6-S (G6S) on chromosome 12q14 (Sanfilippo D; MPS IIID), galactose 6S (Gal 6S) on chromosome 16q24 (Morquio A; MPS IVA), HSulf1 (no OMIM entry) on chromosome 8q13.2-13.3 (isolated disease unknown), and HSulf2 (no OMIM entry) on chromosome 20q13.12) (isolated disease unknown). The residual activity of each enzyme is variable as are the clinical manifestations and severity.
Loss of both intellectual and motor functions is consistent; coarse features, visceromegaly, skeletal anomalies, ichthyosis, and chondrodysplasia punctata are more variable manifestations. Pathologic features reflect storage of sulfatides and mucopolysaccharides.150
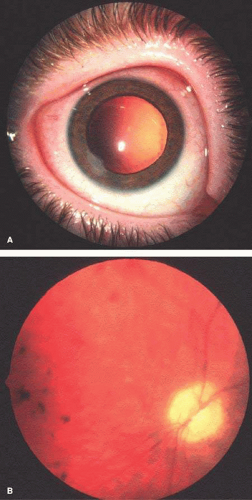 Fig. 4. Multiple sulfatase deficiency. Lens and posterior pole of eye in an affected patient. a: Distinctive peripheral lens opacity may be due to storage material in nucleated lens cells. A superficial corneal scar, related to exposure is evident inferiorly. b: Profound optic atrophy and retinal degeneration with retinal pigment epithelial changes is evident. Although the cataract is not visuallysignificant, the vision is profoundly reduced by the retinal degeneration and optic atrophy. |
Chondrodysplasia punctata, X-linked recessive (OMIM #302950)
The X-linked recessive form of chondrodysplasia punctata is caused by mutations of the arylsulfatase E (ARSE) (OMIM *300180) gene on chromosome Xp22.3; multiple mutations have been described.151
In addition to stippling of the epiphyses, affected males have failure to thrive, developmental delay, and unusual facies.152,153,154
Although reported, cataracts are not common.155
Galactosylceramide lipidosis (Krabbe disease) (OMIM #245200)
Reduced activity of the galactosylceramide β-galactosidase (galactosylceramidase) enzyme is caused by mutations of the gene (GALC) (OMIM *606890) on chromosome 14q25-31. Krabbe disease (galactosylceramide lipidosis; globoid cell leukodystrophy—GLD; GCL) is characterized by a failure of myelinogenesis with loss of myelin-forming cells. The early name for the disease, globoid cell leukodystrophy, reflects the development of globoid cells within the brain caused by accumulation of galactosylceramide156 around cerebral blood vessels. The disorder is inherited in an autosomal recessive pattern and many mutations have been described.157,158,159,160 Diagnosis is made by enzyme activity or DNA sequence analysis of the gene; prenatal diagnosis has been performed successfully.161
Clinically, there are three forms: infantile, later onset, and adult. In the classic form, onset is approximately age 6 months with loss of milestones, visual inattentiveness, hearing loss, and spasticity; the psychomotor deterioration progresses and hypotonia develops. Death usually occurs by age 2 years. The later-onset form accounts for up to 15% of cases; onset is from 15 months to late teenage years. The onset is usually with gait disturbance and psychomotor deterioration. Phenotypic variability occurs even within families162,163 and death usually occurs in childhood. The adult onset is uncommon with the onset of progressive gait disorder leading to spastic paraparesis; intellectual deterioration is not a feature.163,164,165
Prosaposin deficiency (OMIM +76801)
Based on a mouse model, saposin A deficiency was postulated as a potential cause of globoid cell leukodystrophy.168 Recently, a disorder similar to galactosylceramide lipidoses (Krabbe disease) was described with a deficiency of saposin A; mutations of the prosaposin gene (OMIM +76801) have been identified.82,169,170,171 The prosaposin encodes a glycoprotein, which is the precursor of all four saposins.
Clinically, profound neurologic impairment is evident at birth, and death occurs in infancy.170,171 An onset in childhood with seizures and progressive neurologic deterioration have been reported.172
Other Lipidoses
Niemann-Pick, types C1 and D (NPC1) (Niemann-Pick, Nova Scotia type) (OMIM #257220)
Niemann-Pick types C1 and D have similar clinical features to the Niemann-Pick types A and B and are inherited in an autosomal recessive pattern. Mutations of the gene NPC1 (OMIM *607623) on chromosome 18q11-q12, causes both Niemann-Pick types C1 and D; the abnormal protein product causes dysfunction of intracellular trafficking of LDL cholesterol.173 NPC1 encodes a glycoprotein with multiple transmembrane domains with a conserved sterol-sensing domain (SSD), essential for functioning; the protein is a permease, acting as a transmembrane efflux pump.174 The NPC1 protein is located within the endosomal membrane and interacts with lysosomes and Golgi apparatus.175,176 Binding between NPC1 and azocholestanol mediates sterol binding177 and a mutation of this region causes NPC.178 Other DNA regions also are essential for functioning,179,180 and most patients with Niemann-Pick disease type C have mutations of this gene (NPC1).180,181,182,183,184,185,186 Three of the mutations are the most common.182,183,187 The previously-described Niemann-Pick type D, more common in individuals with Acadian Nova Scotian origin, is caused by a mutation of NPC1.188 Clinically and biochemically, there is considerable variation, but these types cannot be differentiated reliably from Niemann-Pick type C2. In types C1, C2, and D, a mixture of lipids, primarily cholesterol and glycosphingolipids, accumulate in lysosomes and endosomes.177,189
Clinically, individuals with mutations of the NPC1 gene, most commonly, have hepatosplenomegaly and progressive neurological dysfunction with varying ages at onset and varying courses.
The ocular features of type C include vertical gaze dysfunction shown to be caused by lesions in the posterior commissure and neuronal loss in the rostral interstitial nucleus of the medial longitudinal fasciculus (MLF).190,191 In one case, optic nerve pallor and perimacular gray discoloration were observed and, on pathologic examination, “pleomorphic membranous inclusions” were found within the conjunctival fibrocytes, vascular endothelial cells and pericytes, keratocytes, lens epithelium, retinal ganglion cells, retinal pigment epithelium, fibrocytes in the uveal tract, and optic nerve fibrous astrocytes.192
The diagnosis of NPC can be made by conjunctival biopsy54 or mutational analysis.
Niemann-Pick, type C2 (OMIM #607625)
NPC2 (HE1) (OMIM *601015) (human epididymis 1) encodes a lysosomal cholesterol-binding glycoprotein with several regions essential for secretion from the lysosome.193 This gene has been mapped to chromosome 14q24 and, when mutated,185,194,195 accounts for a lower percentage of Niemann-Pick type C cases196; the disease is rarer than Niemann-Pick type C1 and is inherited in an autosomal recessive pattern. A mixture of lipids, primarily cholesterol and glycosphingolipids, accumulate in lysosomes and endosomes.177,189
The disease caused by HE1/NPC2 mutations result in a wide spectrum of signs and symptoms. Patients with Niemann-Pick type C2 have significant pulmonary fibrosis.197
Clinically, age of onset varies considerably but is usually early in life with liver dysfunction and neurological deterioration; respiratory disease and neurologic involvement is prominent in the NPC2/HE1.196 In the early onset form, death occurs between the ages of 6 months and 4 years from respiratory failure; some patients have neurological degeneration.183 Less commonly, onset is later in life with visceromegaly.198
Vertical supranuclear ophthalmoplegia is a feature.198
Neuronal Ceroid Lipofuscinoses
The neuronal ceroid lipofuscinoses (NCL) are a group of disorders in the category of liposomal proteinoses199; most are inherited as autosomal recessive traits. There are at least seven disorders and, collectively, the incidence is 1 in 10,000 in the United States and northern Europe.200,201 All have abnormal lysosomal function with accumulation of storage of autofluorescent material in a granular, curvilinear, or fingerprint patterns on electron microscopy. The diseases were originally categorized by the appearance of the storage material by electron microscopy and the eponym Batten disease has been applied to several forms, usually the juvenile form; subsequently, categorization was by age of onset and the names became more specific. The genes for NCL1, 2, and 3 have been identified and all are lysosomal. Electron microscopy of conjunctiva is the easiest format for screening, although white cells and skin or rectal biopsy may be studied. Mutations are expressed in lysosomes (TSD, CLN1/PPT1, CLN2/TTPI, CLN3, and CLN5) and endoplasmic reticulum (CLN6); one type is caused by mutations in a protein that recycles between the endoplasmic reticulum and the endoplasmic reticulum Golgi intermediate compartment (CLN8). Mutations are collated in the NCL Mutation Database (http://www.uc.ac.uk/ncl). Prenatal diagnosis is feasible by electron microscopy202 or gene sequencing.
Infantile neuronal ceroid lipofuscinoses (CLN1) (Hagberg-Santavuori) (OMIM #256730)
The infantile form of neuronal ceroid lipofuscinoses Hagberg-Santavuori primarily affects the Finnish population and is rarely found in other groups. The defect is in the gene palmitoyl protein thioesterase (PPT1) (OMIM *600722) on chromosome 1p32 encoding for the lysosomal protein of the same name. Saposins A and D accumulate in lysosomes.203
The disease begins with a loss of intellectual and motor milestones in the first year of life. The disease progresses relatively rapidly with hypotonia, visual impairment, myoclonic epilepsy, and ataxia. Ultimately, microcephaly with brain atrophy occurs.204
Visual inattentiveness occurs early in the disorder. Retinal degeneration with hypo- and hyperpigmentation develops.205,206 The electroretinogram is abolished and, by histopathology, there are reduced cell numbers in all retinal layers, including the inner nuclear layer, and autofluorescent lipofuscin granules are present.207 Characteristic posterior polar cataracts are a feature of the disease (Fig. 5a).205 The reasons for visual impairment include central nervous system deterioration and both retinal degeneration and optic atrophy (Fig. 5b).
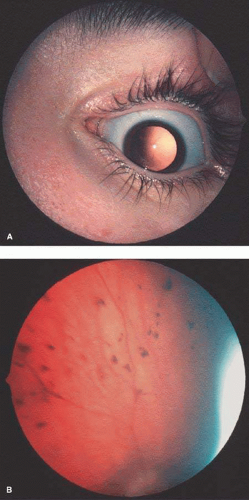
Fig. 5. Hagberg-Santavuori disease. Lens and posterior pole in an affected child. a: A distinctive, star-like cataract is present at the level of the posterior capsule. b: The retina is degenerated with significant pigmentary changes.
Prenatal diagnosis is feasible with gene sequencing.208
Late infantile neuronal ceroid lipofuscinoses (CLN2) (Jansky-Bielschowsky) (OMIM #204500)
The late infantile form of the ceroid lipofuscinoses, Jansky-Bielschowsky disease, occurs in all populations. The defect is in the gene encoding tripeptidyl peptidase I (TPP-I; CLN2) (OMIM #607998) on chromosome 11p15.5.209 The protein product is a lysosomal serine protease that removes tripeptides from the free-N termini of small polypeptides.210 Adenosine triphosphatase (ATP) synthase, subunit c, accumulates in lysosomes.211,212 The disease usually begins with seizures between the ages of 2 and 4 years. Subsequently, the child has progressive loss of intellectual and motor milestones with visual inattentiveness. Ultimately, cerebral and cerebellar atrophy occurs.213
Juvenile neuronal ceroid lipofuscinoses (CLN3) (Spielmeyer-Vogt) (OMIM #204200)
The juvenile form of the neuronal ceroid lipofuscinoses, Spielmeyer-Vogt disease, has been reported in many populations. The gene (CLN3) encodes the CLN3 protein (OMIM *607042) in the lysosomal transmembrane219,220 and has been mapped to chromosome 16p12. ATP synthase subunit C and saposins A and D accumulate in the lysosomes.211,212 Less commonly, mutations of the gene palmitoyl protein thiolesterase (PPT1) for CLN1 (Hagberg-Santavuori disease) (OMIM *600722) causes a form that is similar to the juvenile form of NCL.221
The disease begins with complaints of visual impairment between the ages of 5 to 10 years; frequently, the diagnosis of fictitious vision loss is made. Subsequently, a diagnosis of juvenile macular degeneration or Stargardt macular dystrophy is made when macular changes are evident. Thereafter, behavioral changes usually occur with decreased attention span; neurologic manifestations usually begin within 3 years of the onset of visual impairment.222 Eventually, seizures, ataxia, and progressive loss of intellectual skills and rigidity ensue. The patient becomes vegetative by the mid- to late teens. Cerebral and cerebellar atrophy occur.223
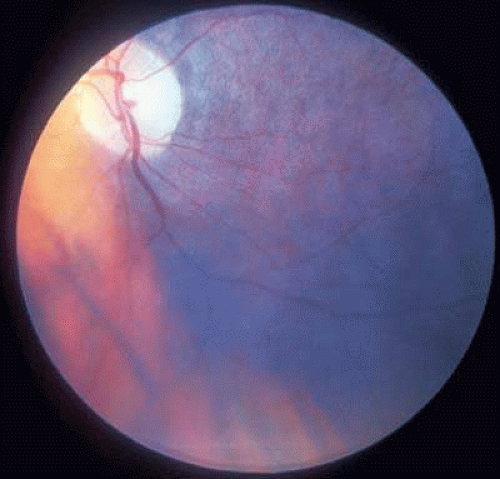 Fig. 6. Speilmeyer-Vogt disease. Macular degeneration pigment epithelial changes in an affected child. |
Adult neuronal ceroid lipofuscinoses (Kufs) (CLN4) (OMIM #204300)
The adult form of the neuronal ceroid lipofuscinoses, Kufs disease, occurs in all populations usually as an autosomal recessive disease (OMIM #204300)228; less commonly, it is inherited as an autosomal dominant trait (OMIM %162350).229,230,231,232,233 In at least one family with an autosomal recessive form, the gene for CLN1 palmitoyl protein thioesterase (PPT1) (OMIM *600722) was mutated and the basis for the disease.234 Other genes have not been identified.
The clinical features of the disease or diseases are more varied than the other forms. Onset of progressive myoclonic seizures or dementia occurs between adolescence and 30 years of age. Ataxia and facila dyskinesias ensue.235,236
Ncl Variants (CLN5, CLN6, CLN7, CLN8)
Several NCL variants have been described and mapped to different chromosomes.
CLN5 (OMIM #256731)
The CLN5 gene (OMIM *608102) has been mapped to chromosome 13q22242 and identified.243,244 The gene is a soluble glycoprotein involved in “trafficking” and is localized to the lysosome.245 Mitochondrial ATP synthase subunit c is the primary stored protein.
CLN6 (OMIM #601780)
CLN6 (EJNCL) has similar features to the juvenile form with loss of vision early in the disease. This form has been mapped to chromosome 15q21-23209,248 and the gene (CLN6) (OMIM *606725) is a transmembrane protein in the endoplasmic reticulum.201,249,250,251 Some families from Turkey with a clinical form similar to CLN7 have a mutation in this gene.252
The onset of symptoms occurs early in life between the ages of 1 and 5 years.209,248 Loss of vision occurs early in the disease, and progressive neurological deterioration ensues.
Retinal degeneration occurs between the ages of 3 to 6 years, and an abnormal electroretinogram is a feature.249
CLN7 (OMIM #600143) and CLN8 (OMIM #600143)
CLN7 has been described in the Turkish population and is similar to the late infantile form (CLN2). CLN8 in the Finnish population is characterized by seizures and progressive mental retardation.253,254 Mitochondrial ATP synthase subunit c is the primary stored protein. Mutations in the CLN8 gene (OMIM *607837) have been found in the Finnish CLN8 patients.255 Some of the Turkish families have a mutation in the CLN8 gene,256 and visual loss has been described. Other Turkish families have mutations in the CLN6 gene.252 However, genotype-phenotype correlation has not been reported. Thus, the Finnish CLN8 and the Turkish CLN7 may be allelic (the same disorder).257,258 The CLN8 protein recycles between the endoplasmic reticulum and the endoplasmic reticulum Golgi intermediate compartment.259
Peroxisomal Disorders
The peroxisomal disorders are a group of diseases in which gene defects disrupt peroxisomal functions. The defects are divided into peroxisome biogenesis diseases (PBD) and single protein/enzyme disorders. Excellent summaries of the mechanisms and features are available.260,261 The pathways are diverse, complex, and interdigitate. The multiple genes encode ether-phospholid biosynthesis, fatty acid β-oxidation, fatty acid α-oxidation, glyoxylate detoxification, biosynthesis of cholesterol and dolichol, pipecolic acid degradation, biosynthesis of docosahexaenoic acid, hydrogen peroxide metabolism, and amino acid metabolism. All are inherited as autosomal recessive traits except X-linked adrenoleukodystrophy. Numerous genes are expressed in peroxisomes, and the organelle directs multiple pathways involving fatty acids and amino acids; peroxisomes divide after reaching a critical size. Some genes are expressed in mitochondria and/or endoplasmic reticulum. At least 17 different disorders have been identified. The disorders of peroxisome biogenesis include Zellweger syndrome, neonatal adrenoleukodystrophy, infantile Refsum disease, hyperpipecolic acidemia, and rhizomelic chondrodysplasia punctata type 1. The single peroxisomal enzyme and protein disorders include X-linked adrenoleukodystrophy, acyl-CoA oxidase deficiency (pseudo-neonatal ALD), D-bifunctional protein deficiency, 2-methylacyl-CoA racemase deficiency, rhizomelic chondrodysplasia punctata type 2 (DHAPAT deficiency), rhizomelic chondrodysplasia punctata type 3 (alkyDHAP synthase deficiency), Refsum disease (phytanoyl-CoA hydroxylase deficiency), hyperoxaluria type 1, glutaric academia type 3, mevalonate kinase deficiency, acatalasemia, and mulibrey nanism. Clinically, the disorders have common features including craniofacial abnormalities and progressive loss of psychomotor skills. Those with reported ophthalmologic manifestations are described below.
Peroxisome Biogenesis Disease
The peroxisomal biogenesis disorders include the group of Zellweger syndromes; some forms are milder and include neonatal adrenoleukodystrophy and infantile Refsum disease. The mutated genes are termed PEX and the protein products peroxins. There are multiple genes in this group and mutations cause cerebrohepatorenal (Zellweger syndrome), neonatal adrenoleukodystrophy, infantile Refsum, and hyperipipecolic acidemia. Gould and Valle262 formulated the current organizational model, and there is considerable clinical overlap among the groups. Usually patients with Zellweger syndrome die before 1 year of age, and those with neonatal adrenoleukodystrophy usually die in late infancy. Patients with infantile Refsum disease have variable hypotonia and psychomotor retardation; they do not have the characteristic defect in neuronal migration (as occurs in Zellweger syndrome). Neonatal seizures are evident in both Zellweger syndrome and neonatal adrenoleukodystrophy. Wanders263 describes the categorization and features of peroxisomal diseases.
Visual impairment was common in a mixed cohort of patients with survival beyond 1 year (excluding the Zellweger phenotype). Most patients had a mutation of the PEX1 gene. Some affected individuals had significant visual impairment at birth with abnormal electroretinograms. Cataracts, abnormal pursuit, and retinal degeneration were evident in this group.264
Cerebrohepatorenal (Zellweger) syndrome (OMIM #214100)
The cerebrohepatorenal syndrome of Zellweger was the initial disease identified as a peroxisomal disorder265 and the clinical features were described in the 1960s.266 The disease is caused by an enzyme deficiency called generalized peroxisomal enzyme deficiency, where peroxisomes are absent or diminished in number.267 Many PEX genes cause cerebrohepatorenal syndrome, if mutated.263 The gene PEX1268 (OMIM +602136), on chromosome 7q21-q22, encodes a protein (Pexlp) of the family AAA ATP-ases and is the most common basis.269,270,271 This protein is a member of complementation group 1. Mutations of the PEX3 gene 272,273,274 (OMIM +603164), a member of complementation group 12 on chromosome 6q23-q24; PEX16275,276 (OMIM +603360), a member of complementation group 9 on chromosome 11p12-p11.2; PEX19 (OMIM +600279),277 a member of complementation group 19 on chromosome 1q22; PEX26278,279 (OMIM +608666), a member of complementation group 8 on chromosome 22q11.21; PEX10280,281,282,283 (OMIM +602859), a member of complementation group 7 on chromosome 1; PEX6284,285,286,287,288 (OMIM +601498), a member of complementation group 4 on 6p21.1; PEX2289 (OMIM +170993), in complementation groups 5 and 10290 on chromosome 8q21.1; PEX14291 (OMIM *601791), in complementation group K292 on chromosome 1p36.2; PEX13262,293 (OMIM +601789), a member of complementation group 13294 on chromosome 2p15; and PEX5295 (OMIM *600414), in complementation group 2 on chromosome 12p13.3 may cause the disease. Mutations of the PEX10 also may cause neonatal adrenoleukodystrophy.281 A more severe form of neonatal adrenoleukodystrophy or Zellweger syndrome is caused by mutations in the PEX12296 (OMIM +601758) gene on chromosome 17, one of the complementation group 3 that encodes a protein (PEX12) containing two transmembrane domains and a zinc-binding domain. It interacts with other proteins of the peroxisomal protein import machinery.297,298,299,300,301 Mutations of PEX26 and PEX1 can also cause neonatal adrenoleukodystrophy or infantile Refsum syndrome269,270,271,278,279; mutations of PEX13 and PEX5 can cause neonatal adrenoleukodystrophy.295,297
Clinically, variable neurodevelopmental delay, liver disease, perceptive deafness, and retinopathy are common to these peroxisomal biogenesis disorders. Affected patients have a high forehead, large fontanelles, epicanthus, a low and broad nasal bridge, micrognathia, redundant skin folds, external ear and feet deformities, and campodactyly. All patients have progressive psychomotor retardation that is evident early in life. Neonatal seizures with hypotonia and hypoflexia are a consistent feature. Hepatomegaly with subsequent cirrhosis and renal cysts are common.302,303,304 Chondrodysplasia punctata with stippling of the epiphyses is evident.
Most reports of the ocular features of Zellweger syndrome predate the genetic and biochemical understanding of the disease. Reported ocular features include corneal clouding, cataracts, glaucoma, Brushfield spots, and retinal degeneration.263 Cataracts occur in affected individuals and may occur in carrier parents.305,306 Retinal dystrophy is a consistent feature of cerebrohepatorenal syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease.263,305,307,308
Neonatal adrenoleukodystrophy (OMIM #202370), infantile Refsum syndrome (OMIM #266510) and rhizomelic chondrodysplasia punctata type 1 (PCDP1) (OMIM #215100)
As in Zellweger syndrome, the genetic bases of neonatal adrenoleukodystrophy (NALD), infantile Refsum syndrome (IRD), and rhizomelic chondrodysplasia punctata type 1 (RCDP1) are mutations of the PEX genes. Neonatal adrenoleukodystrophy309 (NALD) and infantile Refsum syndrome310,311 are milder variants of Zellweger disease and in the category of peroxisomal biogenesis disorders. The two differ clinically and each has several reported mutations of PEX genes. Rhizomelic chondrodysplasia punctata type 1 is a dwarfing syndrome associated with stippling of the epiphyses.
Ocular features in a cohort of 31 patients with clinical features of a generalized PBD, excluding cerebrohepatorenal (Zellweger) syndrome, included high myopia, nystagmus (39%), strabismus (55%), cataract (10%), retinal degeneration (84%), and optic atrophy(55%).264 Histopathology has shown anterior subcapsular cataracts and macular edema; retinal changes suggestive of retinal degeneration were observed.307
Neonatal adrenoleukodystrophy (OMIM #202370)
Clinically, neonatal adrenoleukodystrophy (NALD) is characterized by neonatal hypotonia, seizures, and some central nervous system anomalie malformations; it differs from the X-linked form in the central nervous system malformations such as heterotopia and microphachygyria.309,311,312 The X-linked form (X-ALD) is due to the loss of function of a peroxisomal protein whereas neonatal adrenoleukodystrophy is a peroxisomal biogenesis disorder.
For neonatal adrenoleukodystrophy, the PEX1 gene is mutated most commonly264 with some residual activity.269 Other genes, which when mutated cause this phenotype, include PEX26,278 PEX10,282,283 PEX6,288 PEX13,297 PEX5,313 and PEX12.297,298 The ocular features include degeneration of photoreceptor cells, and vitreous opacities have been documented.314
Infantile Refsum syndrome (OMIM #266510) (infantile phytanic acid storage disease)
In infantile Refsum syndrome, the PEX1 gene is most commonly mutated264 and associated with some residual activity.269 However, mutations in PEX26,278 PEX10,282,283 PEX6,288 PEX5,313 PEX2,315 and PEX13297,298 have been reported.
The disease is characterized by progressive psychomotor retardation, dysmorphic features, liver disease, deafness, and retinal degeneration.310,311,316
In addition to the retinal degeneration, cataracts have been described.317
Chondrodysplasia Punctata
Chondrodysplasia punctata (CDP) is a group of bone dysplasias characterized by stippling of the epiphyses. The diagnosis is suggestive based on the radiological findings of stippled epiphyses and is confirmed by other clinical features; in some cases, biochemical and DNA testing are the bases. Autosomal dominant, X-linked dominant, X-linked recessive, and autosomal recessive forms are known. At least three types of autosomal recessive chondrodysplasia punctata (RCDP) forms have been described. The common features include psychomotor delay, craniofacial abnormalities, cataracts, rhizomelia, and symmetrical dwarfism, similar to the clinical features of Zellweger syndrome patients.
Rhizomelic chondrodysplasia punctata, type 1 (OMIM #215100) (PCDP1)
Patients with rhizomelic chondrodysplasia punctata type 1 have a defect of the PEX7 gene (OMIM *601757) on chromosome 6q22-q24318,319,320 and the protein product functions as a peroxisomal targeting signal (PTS1) protein.321 A mouse model has defective neural migration.322
The clinical features are similar to other forms of rhizomelic chondrodysplasia punctata with stippled epiphyses, symmetric dwarfism, ichthyosis, and severe mental retardation.323,324 Phytanic acid becomes elevated.
Single Peroxisomal Enzyme and Protein Disorders
Rhizomelic chondrodysplasia punctata type 2 (RCDP2) (DHAPAT deficiency) (OMIM #222765)
Rhizomelic chondrodysplasia punctata type 2 is caused by a deficiency of the protein dihydroxyactonephosphate acyl-transferase (DHAPAT; DAPAT); the DHAPAT gene326 (OMIM *602744) is on chromosome 1q42. The protein is localized entirely within peroxisomes and a deficiency results in an inability to synthesize ether-phospholipids, including plasmalogen.327 Several mutations have been reported.328
Although the clinical features are similar to the other forms of rhizomelic chondrodysplasia punctata,326,329 not all patients have rhizomelia or cataracts.327,329,330,331,332,333,334
Rhizomelic chondrodysplasia punctata type 3 (alky-DHAP synthase deficiency) (OMIM #600121)
RCDP type 3 is due to isolated alky-DHAP synthase deficiency, a peroxisomal protein.336 The gene for alkyldihydroxyacetonephosphate synthase (alkyl-DHAP synthase) (AGPS) (OMIM *603051) is on chromosome 2q31, and several mutations have been described.328,337 Clinically, affected individuals have the RCDP features.321,336
The ocular features have not been described.
X-linked adrenoleukodystrophy (OMIM #300100)
X-linked adrenoleukodystrophy (X-ALD) is caused by mutations of the ABCD1 (OMIM *30371) gene, on Xq28 that encodes a transmembrane regulator protein in the ATP-binding cassette half-transporter superfamily.338,339,340 The degradation of very long chain fatty acids is in the β-oxidation pathway and the protein product of the ABCD1 gene is involved in the transport of the C26:0-CoA ester across the peroxisomal membrane and β-oxidization.261 Accumulations of saturated very long-chain fatty acids, particularly hexocosenoic acid (C26:0) in peroxisomes and the endoplasmic reticulum is the basis of the disease. Many mutations have been identified,341,342,343,344 and a mutational database is maintained (http://www.x-ald.nl).
The phenotype varies considerably. In the childhood variety, the onset of behavioral problems is between 3 and 10 years of age with rapidly progressive cerebral demyelinization.345 Progressive loss of intellectual and motor milestones occur; visual inattentiveness is a relatively early feature. Death usually occurs within a few years. The adolescent form has a later onset and the adult form usually presents as schizophrenia. Some patients present with adrenocortical dysfunction initially. There is poor genotype-phenotype correlation.320 Heterozygote females may develop cerebral demyelination.346 Most affected boys have adrenocortical insufficiency.
Visual impairment is a prominent clinical sign. The ocular features include loss of vision and visual inattentiveness related to central nervous system and retinal ganglion cell demyelination, cataracts, strabismus, macular pigmentary disturbances, and optic atrophy; optic nerve hypoplasia has been described.216
Acyl-CoA oxidase deficiency (pseudoneonatal adrenoleukodystrophy) (OMIM #264470)
Acyl-CoA oxidase deficiency (pseudoneonatal adrenoleukodystrophy) (NALD) (OMIM *601651) is an autosomal recessive disorder described in very few patients. The gene for peroxisomal straight-chain acyl-CoA oxidase (ACOX1) (OMIM *609751) is on chromosome 17q25 and the protein product is the first and rate-limiting enzyme in the peroxisomal fatty acid beta-oxidation pathway. Several mutations have been reported.348,349
Clinically, affected individuals display neonatal hypotonia, seizures, and delayed psychomotor development with progressive loss of developmental milestones after age 2 years.350
A retinopathy has been described but not characterized.351
D-bifunctional enzyme deficiency (OMIM #261515)
D-bifunctional protein deficiency (DBP) is a rare autosomal receive disorder in the pathway of peroxisomal fatty acid oxidation. The enzyme D-peroxisomal bifunctional catalyzes the second and third steps of peroxisomal β-oxidation of fatty acids; the enzyme consists of three functional units. Type 1 individuals have deficiency of the hydratase and dehydrogenase units; type 2 have isolated deficiency of the hydratase unit, and type 3 have deficiency of the dehydrogenase unit.352 Multiple mutations of the DBP gene (DBP) (17-HSD17B4) (OMIM *601860), on chromosome 5q2, have been reported.353,354,355
Clinically, neonatal hypotonia, seizures, and macrocephaly are evident.356,357,358 Most affected children die by the age of 2 years.
Alanine-glyoxylate aminotransferase deficiency (primary hyperoxaluria type 1) (OMIM #259900)
In most cases, primary hyperoxaluria type 1 is caused by a deficiency of alanine-glyoxylate aminotransferase and mutations of the alanine-glyoxylate aminotransferase gene (AGXT)362 (OMIM *604285), on chromosome 2q36-q37. The protein is in the peroxisomes of the liver. Multiple mutations have been reported.363,364,365
Clinically, individuals with primary hyperoxaluria type 1 have recurrent calcium oxalate nephrolithiasis and progressive renal disease.
Refsum disease (OMIM #266500)
Refsum disease (RD), an autosomal recessive disorder, is caused by deficiency of the peroxisomal enzyme phytanoyl-cohydroxylase (PhyH), which catalyzes the first step in α-oxidation of phytanic acid, an exogenous branched chain fatty acid; phytanic acid accumulates in tissues. The gene (PHYH) (OMIM *602026) is on chromosome 10pter-p11.2 and is mutated in most patients with this disease372,373; most mutations are personal.374,375 Less commonly, patients with the clinical features of Refsum disease have a mutation in the PEX7 (OMIM *601757) on chromosome 6q22-q24.318,375
The clinical features of Refsum disease include cerebellar ataxia and a slowly progressive polyneuropathy. Patients develop significant visual impairment early in the disease due to retinal degeneration. Other manifestations include nerve deafness, cardiomyopathy, or ichthyosis.
Amino Acid Metabolism Disorders
Hyperphenylalaninemias
Although the vast majority of hyperphenylalaninemias are phenylketonuria, enzyme deficiencies in dihydrobiopterin synthesis or mutations in the gene encoding dihydropteridine reductase may cause the disease.378
Phenylketonuria (OMIM +261600)
The prototype amino acid metabolism disorder is phenylketonuria (PKU), which was originally described in 1953 by Jervis.379 The enzyme defect in this autosomal recessive disorder is in the phenylalanine hydroxylase protein, the gene (PKU) for which is on chromosome 12q24.1. Mutations in the gene cause elevation of phenylalanine with some correlation of the severity of enzyme dysfunction and phenylalanine intolerance with the genotype.380,381
Untreated infants develop significant mental retardation, eczema, hypopigmentation, and other neurologic disease.
Although ocular disease is usually limited to hypopigmentation, treated patients may have impaired contrast sensitivity.382,383 Premature cataracts may be related to self-abusive behavior.382
Tyrosine Metabolism
There are three types of tyrosinemia; ocular manifestations are evident in both type I and type II (see entries). Type III is caused by mutations of the HPD gene, encoding 4-hydroxyphenylpyruvate dioxygenase, on chromosome 12q24-qter; individuals with this type do not develop ocular abnormalities.386
Tyrosinemia type I (hepatorenal tyrosinemia) (OMIM +276700)
Hepatorenal tyrosinemia, tyrosinemia, type I, is caused by mutations of the gene fumarylacetoacetate hydrolase (FAH) (OMIM +276700) on chromosome 15q23-q25; the protein product, fumarylacetoacetate hydrolase, is an enzyme in the cytosol and reduced activity results in accumulation of succinylacetone and tyrosine. A single point mutation accounts for most of the disease in Quebec, Canada, a region of clustering.387
In the acute infantile form, liver failure occurs. In the chronic form, the liver failure is chronic with development of cirrhosis and renal disease; neurologic crises occur.388
Corneal opacities are common.389
Tyrosinemia type II (oculocutaneous tyrosinemia) (OMIM +261600)
Oculocutaneous tyrosinemia, tyrosinemia II, or Richner-Hanhart syndrome is an autosomal recessive disorder caused by deficiency of tyrosine aminotransaminase (TAT) (OMIM +276600), on chromosome 16q22.1-q22.3. Tyrosine accumulates and the plasma tyrosine is elevated.
Clinical manifestations include blisters on the fingertips and thenar eminences that progress to form painful hyperkeratotic plaques on the palms and soles of the feet. Some patients have developmental delay.391,392
Alkaptonuria (OMIM #203500)
Alkaptonuria (AKU), or onchronosis, is an autosomal recessive disorder of homogentisic acid metabolism. The disease became historic through the studies of Sir Archibald Garrod in 1902.398 Mutations of the gene HGD (OMIM *607474), on chromosome 3p21-q23, cause deficiency of homogentisate 1,2-dioxygenase, an enzyme that metabolizes homogentisic acid and the disease. Homogentisic acid accumulates and oxidizes resulting in a pigmented polymer. Many mutations have been reported.399,400 The disorder is more common in Slovakia and the Dominican Republic.401,402,403
Clinically, urine turns dark brown after exposure to oxygen. Affected individuals develop arthritis and/or aortic and mitral valve stenosis.404
Pigmentation is noticeable in sclera and cartilage of the ears, usually by the second or third decade.405
Disorders of Sulfur Amino Acid Metabolism
The diseases of sulfur amino acid metabolism involve methionine, homocysteine, cystathionine, and cysteine; the B12 metabolism disorders also cause diseases of the sulfur amino acids. Methionine is an essential amino acid and homocysteine is essential for the transsulfuration pathways. The term homocystinuria refers to excess homocystine in the urine and represents a group of diseases. Currently, these diseases should be categorized by enzyme defect.
In addition to cystathionine beta-synthase deficiency, the most common form, other defects that cause homocystinuria include abnormalities in vitamin B12 metabolism; deficiency of N(5,10)-methylenetetrahydrofolate reductase; intestinal malabsorption of vitamin B12; vitamin B12-responsive homocystinuria, cblE type; methylcobalamin deficiency, cblG type; vitamin B12 metabolic defect, type 2; cobalamin disorders of both methylmalonic aciduria and homocystinuria (cblC, cblD and cblF); and transcobalamin II deficiency.
Homocystinuria (cystathionine β-synthase deficiency) (OMIM +236200)
Homocystinuria is usually caused by reduced activity of cystathionine β-synthase and is an autosomal recessive disorder; both homocystine and methionine accumulate and are excreted in the urine. The gene (CBS) is on chromosome 21q22.3 and many mutations have been reported.407,408,409 Some mutations result in defective enzymes in which the activity is increased by B6 administration.
Clinically, affected individuals have the body habitus of patients with Marfan syndrome, being tall and slender with scoliosis, pectus excavatum and arachnodactyly, and elongation of the digits. The skin and hair tend to be pale and hypopigmented. Developmental delay, osteoporosis, pancreatitis,410 and spontaneous pneumothorax411 may develop. Importantly, vascular thromboses are more common and life threatening; the risk has been estimated to be approximately 4% per year.412 Developmental delay is usually greater in the B6-unresponsive individuals.412
Ocular features include a progressive lens dislocation due to dissolution of the lens zonules (Fig. 7) that are predominantly composed of fibrillin-1, a protein with high cysteine; homocysteine may bind the cysteines in fibrillin-1. Myopia is common. Glaucoma may occur as a result of dislocation of the lens into the anterior chamber (Fig. 8).413,414 Dislocation of the lens by the age of 10 years was found in 55% of the B6 responsive group and 82% in the unresponsive group412; additionally, methionine restriction initiated early in life prevented mental retardation and slowed the rate of lens dislocation.412 If initiated early in life, B6 may reduce lens dislocation.415 Ocular axial length is significantly increased in individuals with homocystinuria and lens dislocation.416 Lensectomy may be warranted.413
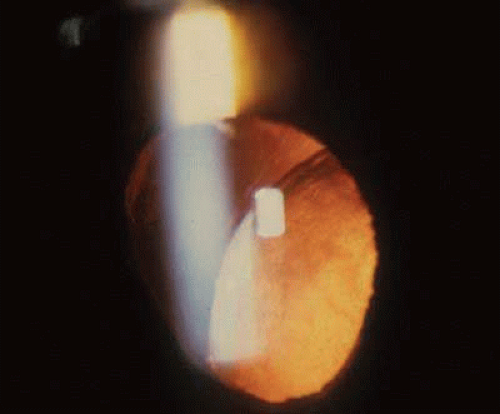
Fig. 7. Homocystinuria. Lens dislocation with dissolution of zonules. (Courtesy of Irene H. Maumenee MD)
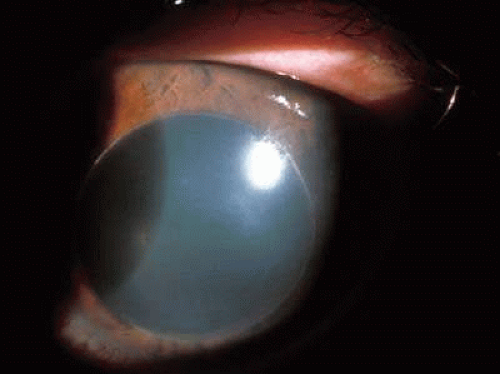
Fig. 8. Homocystinuria. Dislocation of lens into anterior chamber in an affected young adult. (Courtesy of Irene H. Maumenee MD)
Screening and diagnosis are by urine amino acid chromatography. Most state newborn screening programs include homocystinuria; some methodologies might miss the pyridoxine responsive form.417 Treatment includes pyridoxine (vitamin B6). The pyridoxine-responsive forms are associated with a milder phenotype and a better prognosis. Approximately 50% of patients are responsive to pyridoxine.412,418 Treatment with dipyridamole and vitamin C may reduce the risk of thromboembolic phenomenon.419 Low methionine diet has been effective in lowering homocysteine.412
Homocystinuria (OMIM #236250) (5,10-methylenetetrahydrofolate reductase [MTHFR] deficiency)
Accumulation of homocystine with homocystinuria is less commonly caused by a mutation of 5,10-methylenetetrahydrofolate reductase gene (MTHFR) (OMIM *607093), on chromosome 1p36.3. The enzyme catalyzes the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, a cosubstrate for homocysteine remethylation to methionine.
Variability is significant and siblings may have different manifestations while some affected individuals are asymptomatic.420 Clinically, older children and adults may have developmental delay and ataxia. Affected infants may have hypotonia and failure to thrive. One allele predisposes to premature cardiovascular disease421 and neural tube defects.422 Multiple mutations have been reported.423,424
Homocystinuria (methylcobalamin deficiency)
Homocystinuria due to methylcobalamin deficiency occurs in a group of diseases caused by deficiencies of several different enzymes in the cobalamin complementation groups (CblA through H); all are autosomal recessive. Decreased methylcobalamin and adenosylcobalamin results in elevation of homocysteine with homocystinuria and methylmalonic acid. Abnormalities of precellular defects resulting in methylcobalamin deficiency are milder diseases than those of the complementation group for intracellular cobalamin metabolism. Methylcobalamin is the cofactor for the conversion of homocysteine to L-methionine by the enzyme N5-methyltetrahydrofolate: homocysteine methyltransferase (methionine synthase) (OMIM #156570), the gene (MTR) for which is on chromosome 1q43. Adenosylcobalamin is an essential cofactor for the mitochondrial enzyme methylmalonyl-CoA mutase (MUT) (OMIM *609058).
Clinically, megaloblastic anemia is a feature and some forms have methylmalonic acidemia. Features vary considerably but are similar to other forms of homocystinuria.
Methylcobalamin deficiency in CblC (OMIM #277400)
Methylcobalamin deficiency CblC is associated with reduced activity of methylmalonyl-CoA mutase (MUT) (OMIM *609058) and L-methionine by the enzyme N5-methyltetrahydrofolate: homocysteine methyltransferase (methionine synthase) (MTR) (OMIM *156570). The defect is in the gene MMACHC, methylmalonic aciduria cblC type with homocystinuria, on chromosome 1p, that may be involved with energy transduction for cobalamin uptake. Many mutations have been identified, and there is genotype-phenotype correlation; one mutation accounts for 40% of the mutant alleles.427
Both pediatric and adult forms exist. Affected children have normal development and no neurologic symptoms; they have renal disease with proteinuria and hematuria, hypertension, and chronic hemolytic anemia.428 Adults develop a progressive paraplegia, encephalopathy, and neurologic degeneration.427,429
Methylcobalamin deficiency in CblD (OMIM %277410)
The underlying metabolic defect of methylcobalamin deficiency CblD is unknown.
Clinically affected individuals have progressive neurologic deterioration.
Visual inattentiveness is a feature, and the electroretinogram is normal.430
Methylcobalamin deficiency in CblE (OMIM #236270)
Methylcobalamin deficiency due to defects in CblE is, in some cases, due to mutations of the gene encoding methionine synthase reductase (MTRR)431,432 (OMIM *602568) on chromosome 5p15.2-15.3; this gene is distinct from the MTR gene on chromosome 1, which is mutated in CblG disease. Multiple mutations of the MTRR gene have been reported.431,433
Methylcobalamin deficiency in CblF (OMIM %277380)
The underlying metabolic defect of methylcobalamin deficiency CblF is unknown. Very few patients have been reported.
At least one patient had a normal eye exam.437
Methylcobalamin deficiency in CblG (OMIM #250940)
The molecular basis of methylcobalamin deficiency type CblG is the gene MTR (OMIM *156570) encoding methionine synthase (methyltetrahydrofolate homocysteine methyltransferase), located on chromosome 1q43.438 Multiple mutations have been reported.438
Clinically, the disease is similar to CblE with a progressive neurologic deterioration.439 Renal failure may develop.
Ocular features include visual inattentiveness. “Rotating eye movements,” suggestive of nystagmus, have been described.439 The electroretinogram is abnormal with reduced cone and rod amplitudes. Visual evoked responses were abnormal434,440; the authors postulated both photoreceptor and ganglion cell loss caused by microvascular disease.
Sulfocysteinuria (OMIM #272300)
Sulfite oxidase deficiency, a rare autosomal recessive disorder of the degradation pathway of amino acids containing sulfur, is basis of sulfocysteinuria. The gene SUOX (OMIM *606887), on chromosome 12, encodes a protein product with a molybdenum site; the oxidative activity is the terminal degradative reaction.441 Many mutations have been documented.442,443,444,445,446
The clinical manifestations usually include severe neurodegeneration and seizures relatively early in life; death occurs in infancy or early childhood.443,447 Milder forms with a later onset have been described. A similar phenotype occurs as a result of abnormalities of any of the genes in the synthesis of molybdopterin (molybdenum cofactor deficiency; see entry).
Acquired ecoptia lentis is a feature and may occur in the first year of life.443,444,445,447,448,449 As the glycoprotein in zonules has a high concentration of cysteine, zonules are subject to abnormal formation in diseases of sulfur metabolism.450 The mechanism may be related to a decrease in the tissue inhibitor of metalloproteinases and an increase in matrix metalloproteinases.451
Molybdenum cofactor deficiency (OMIM #252150)
Molybdenum cofactor deficiency (COCOD), originally described in 1978,453 is caused by defects in either of the two proteins that synthesize molybdenum cofactor. The gene for molybdenum cofactor synthesis-1 (MOCS1) (OMIM *603707), on chromosome 6p21.3, encodes two enzymes in the pathway, and the gene for molybdopterin cofactor synthase (MOCS2) (OMIM *603708), on chromosome 5q11, encodes two subunits of different sizes.454,455 The third gene, GEPH (OMIM *603930) on chromosome 14q24, in the cycle encodes gephyrin and also causes molybdenum cofactor deficiency.456 Mutations in each of the genes, MOCS1,454,455,457,458 MOCS2,455,457,458 and GEPH,456,457,458 have been reported, primarily in Europe.457
The phenotype is similar to sulfite oxidase deficiency in each of the groups. Clinically, neurodegeneration begins in infancy and there is profound mental retardation.
Reported ocular features include nystagmus, enophthalmos, Brushfield spots, spherophakia possibly related to dissolution of zonules, and lens dislocation.448,459,460 Lens dislocation may occur later in childhood.460 Not all individuals with a mutation of MOSC1 gene have a dislocated lens.458
Prenatal diagnosis has been successful in all three forms.452
Branched-Chain Amino Acids
Maple syrup urine disease (OMIM #248600)
Maple syrup urine disease (MSUD) may be caused by mutations of any one of four genes: branched-chain alpha-keto acid dehydrogenase complex with the α subunit of E1 (BCKDHA) (OMIM *608348) on chromosome 19q13.1-13.2,461 the β subunit of E1 (BCKDHB) (OMIM *248611) on chromosome 6p22-p21,462 dihydrolipoamide branched-chain transacylase (E2) (DBT) (OMIM *248610) on chromosome 1p31,463 and homodimeric dihydrolipoamide dehydrogenase (E3) (DLD) (OMIM *238331) on chromosome 7q31-32464,465; all are inherited in an autosomal recessive pattern and are in the branched-chain alpha-keto acid dehydrogenase (BCKD) complex, which is the catabolic of the branched-chain amino acids, leucine, isoleucine, and valine.
Originally described by Menkes et al.466 in 1954, affected individuals have a neurodegenerative disorder with a urinary odor of maple syrup. The classic, most severe form of the disease has a neonatal onset and is the most common; the intermediate and intermittent forms have the onset of symptoms in childhood. The E3 deficient form is associated with lactic acidosis.
The ocular features include nystagmus, strabismus, ophthalmoplegia, optic atrophy, and cortical “blindness.”467,468
Treatment is dietary and some forms respond to thiamine.
Disorders of Proline and Hydroxyproline
Peptidase D deficiency (OMIM +170100)
Defects of the peptidase D (prolidase) gene (PEPD), on chromosome 19cen-q13.11, alter collagen catabolism.469
Clinically, peptidase D deficiency has variable manifestations. It is characterized by mild mental retardation, facial dysmorphism, recurrent respiratory infections, diarrhea, and dermatitis. Onset can occur at birth or into the third decade.
Ocular features include high myopia.470
Disorders of the Urea Cycle and Ornithine
The disease hyperammonemia may be caused by several enzyme deficiencies and is usually very serious or lethal early in life. Ocular features have not been reported.
Hyperornithinemias
Hyperornithinemia is associated with three gene defects. Deficiency of ornithine transcarbamylase, on chromosome Xp21.1, is associated with hyperammonemia; reduced activity of mitochondrial ornithine transporter (OMIM *603861), on chromosome 13q14, is associated with both hyperammonemia and homocitrullinuria (HHH). Both are mitochondrial proteins, and ocular manifestations have not been reported. Some affected individuals die early in life. Defects in the gene encoding ornithine aminotransferase cause a retinal degeneration termed gyrate atrophy.
Gyrate atrophy (ornithine aminotransferase deficiency) (OMIM +258870)
Hyperornithinemia caused by ornithine aminotransferase deficiency is an autosomal recessive disease. The gene OAT is on chromosome 10q26; however, several pseudogenes (OMIM *311240) are on Xp11-p11.23 and are not known to cause disease.471 The disease is more common in Finland.
Clinically, affected individuals are normal aside from the characteristic retinal degeneration. Biopsy of skeletal muscle has demonstrated abnormalities of type 2 fibers.472,473
The ocular features are distinctive. Affected individuals experience symptoms of retinal degeneration with nyctalopia (night blindness) relatively early in life. The appearance of the retina is unusual with progressively enlarging regions of chorioretinal atrophy that are sharply demarcated (Fig. 9). The features vary considerably among individuals, even with the identical mutation.474
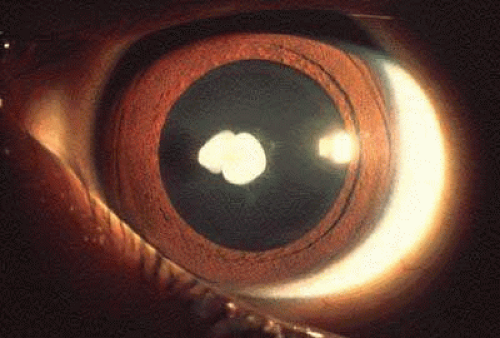
Fig. 9. Gyrate atrophy. Scalloped and sharply demarcated edges to the chorioretinal atrophy in an affected patient. (Courtesy of John Heckenlively MD)
To date, the only available therapy has been dietary arginine restriction relatively early in life, an onerous diet. Slowing of the retinal degeneration has been demonstrated.475
Disorders of Carbohydrate Metabolism
Disorders of carbohydrate metabolism vary in complexity and ocular manifestations.
Galactose Metabolism
Three enzymes convert galactose, a component of lactose present in milk, to glucose-6-phospate, an important source of calories, particularly for infants. All three genes, galactokinase (GALK1) (OMIM *604313) on chromosome 17q24, galactose-1-uridyl transferase (GALT) (OMIM *606999) on chromosome 9p13, and uridine diphosphate galactose-4-epimerase (GALE) (OMIM *60695) on chromosome 1p36-p35 are expressed in many tissues. Deficiency of each is inherited as an autosomal recessive disease. The term galactosemia is usually reserved for the transferase deficiency. Because of the profound mental retardation associated with the transferase deficiency, newborn screening for galactosemia has been legislated in all states in the United States. Urinary testing for sugar is rarely used diagnostically as it is dependant on the methodology (and manufacturer) of the test as well as the time of last milk ingestion; galactosuria is greatest in galactosemia. All deficiencies are definitively diagnosed by red cell enzyme assay.
Galactokinase deficiency (OMIM #230200)
Galactokinase deficiency was initially identified in the mid-1960s.476 The gene GALK1, on chromosome 17q24, encodes the first enzyme in the pathway, and several mutations have been identified.477,478 The frequency is estimated to be 1 in 1,000,000479 except in the Roma Gypsy group where it is more common.480 Affected individuals have elevated galactose.
Generally, affected individuals are asymptomatic with the exception of cataracts. Pseudotumor cerebri has been documented.481 Although classically described as “oil droplet,” the ocular features are acquired cataracts of variable morphology in untreated individuals (Figs. 10 and 11).
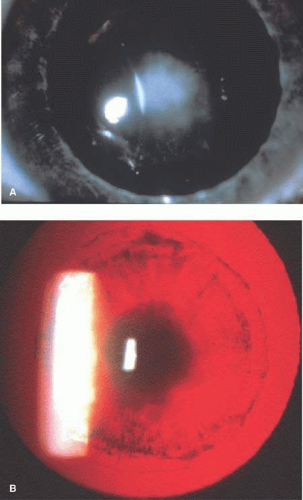
Fig. 10. Galactokinase deficiency. Cataract in a child. (Courtesy of Frederick Wang MD)
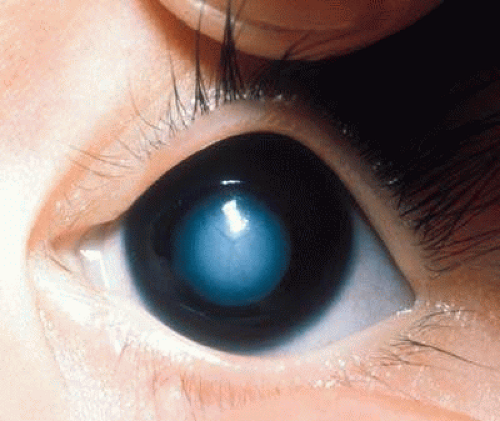
Fig. 11. Galactokinase deficiency. Cataract in child with galactokinase deficiency with (a) direct and (b) indirect illumination. (Courtesy of Irene H. Maumenee MD)
Treatment consists of exclusion of lactose and other sources of galactose from the diet.
Galactosemia (OMIM #230400)
The term galactosemia usually refers to the disease associated with deficiency of galactose-1-uridyl transferase, the gene for which is (GALT) (OMIM *606999) on chromosome 9p13. The frequency is approximately 1:47,000 in the United States.482 Many mutations have been identified and they cluster in ethnic groups.483,484
Clinically, affected infants are normal at birth and usually present with failure to thrive. They develop lethargy, hypotonia, vomiting, diarrhea, and hepatomegaly with jaundice. If untreated, death occurs.
Cataracts develop relatively early in life in untreated, affected individuals (Figs. 12 and 13). Heterozygotes may also develop cataracts prematurely.485
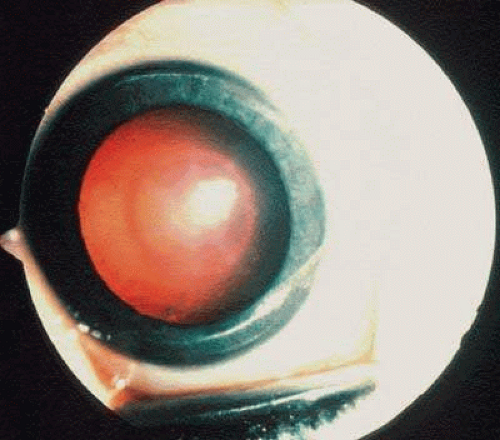
Fig. 12. Galactosemia. Cataract in an infant with galactose-1-uridyl transferase deficiency. (Courtesy of Sherwin J. Isenberg MD)
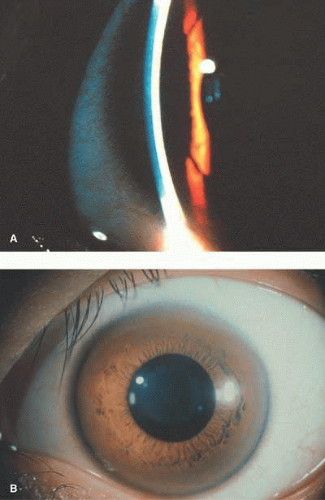
Fig. 13. Galactosemia. Oil droplet cataract in an infant with galactose-1-uridyl transferase deficiency. (Courtesy of Allen Eisenbaum MD)
As the mental retardation is preventable, all states in the United States require testing of newborns. Treatment is exclusion of lactose and other sources of galactose from the diet.
Uridine diphosphate (UDP) galactose-4-epimerase deficiency (OMIM #23035)
Uridine diphosphate galactose-4-epimerase deficiency occurs in a benign and severe form. The enzyme catalyzes two different reactions, both epimerization of UDP-glucose to UDP-galactose and UDP-N-acetylglucosamine to UDP-N-acetylgalactosamine. The gene (GALE) is on chromosome 1p36-p35 and several mutations have been described.486,487,488; there is genotype/phenotype correlation with the mutation.486,488
In the benign form, the infants are normal and identified through neonatal screening programs because of increased erythrocyte galactose-1-phosphate levels. The reduced epimerase activity is limited to the red and white cells; no treatment is given. The severe form is extremely rare and the clinical manifestations are similar to galactosemia.489,490
Unlike the galactokinase and transferase (galactosemia) diseases, affected individuals cannot endogenously synthesize UDP-galactose and treatment is a galactose-restricted diet (as opposed to a galactose-free diet as is necessary in the other forms).493
Glycogen Storage Diseases
Glucose-6-phosphate deficiency (OMIM +30599)
Glucose-6-phosphate deficiency (G6PD) is an enzymatic defect in hexose monophosphate pathway erythrocytes.494,495 The variants are more common in the African, Mediterranean, and Asiatic population.496 The gene (G6PD) is on chromosome Xp28, and deficiencies are inherited in an X-linked recessive pattern. Many mutations have been reported497; a website compilation is available at www.bioinf.org.uk/g6pd/. The high prevalence is due to relative protection from malaria in hemizygotes and heterozygotes.498
Affected individuals may develop diet- or drug-sensitive hemolytic anemia, newborn anemia and jaundice, and/or chronic nonspherocytic hemolytic anemia.499,500
Glucose-6-phosphatase deficiency (glycogen storage disease type Ia, von Gierke disease) (OMIM +232200)
Glucose-6-phosphatase deficiency type 1a (GSD1a) is caused by mutations of the gene (G6PC) encoding glucose-6-phosphatase on chromosome 17q21 and is inherited in an autosomal recessive pattern. The protein is within a complex of transport proteins, and multiple mutations have been reported.503,504,505
Clinically, the disease is a disorder of the liver and kidney associated with hyperlipidemia and xanthoma formation.506,507,508
Glycogen storage disease type Ib (OMIM #232220)
Glycogen storage disease type Ib (GSDIb) is caused by mutations of the gene encoding the glucose-6-phosphate transporter gene (G6PT1) (OMIM *602671)512 on chromosome 11q23. The disease is autosomal recessive, and multiple mutations have been reported.512,513,514,515 The protein is expressed in the endoplasmic reticulum, primarily in liver, kidney, and hematopoetic cells.
Clinically, the disease is similar to the glucose-6-phosphate deficiency.516 Hepatomegaly and enlarged kidneys as well as neutropenia are features of the diseases.
The ocular features include delayed appearance of the choroidal flush on fluorescein angiography, abnormal electro-oculography, and atrophy of the retinal pigment epithelium and choriocapillaris.509
Gluconeogenic Disorders Associated with Lactic Acidosis
Purine Metabolism
Lesch-Nyhan disease (OMIM #300322)
Lesch-Nyhan disease (LNS) is caused by mutations of the gene (HPRT) (OMIM *308000) encoding hypoxanthine guanine phosphoribosyltransferase; the disease is an X-linked recessive, and the gene is on chromosome Xq26-q27.2. Affected individuals have excessive purine synthesis; multiple mutations have been reported.517 A partial deficiency results in the Kelley-Seegmiller syndrome (OMIM #300323).518,519
The characteristic clinical features include mental retardation, spastic cerebral palsy, choreoathetosis, uric acid urinary stones, and self-destructive behavior. Those individuals with the Kelley-Seegmiller syndrome present with gout and kidney stones but rarely the neurologic features.518,519
Ocular features include abnormal eye movements,520 and self-mutilation and retinal detachment and/or enucleation.521
Prenatal diagnosis has been successful.522
Lipoprotein and Lipid Metabolism
High-Density Lipoproteins and Reverse Cholesterol Transport
Lecithin: cholesterol acyl transferase (OMIM #245900; OMIM #136120) deficiency
Lecithin: cholesterol acyl transferase (LCAT) esterifies high-density lipoprotein (HDL) cholesterol in a process called reverse cholesterol transport. Mutations of the gene (LCAT) (OMIM *606967), on chromosome 16q22.1, causes an autosomal recessive disease and are associated with varying degrees of deficiency523,524,525; partial activity results in the “fish-eye” disease (OMIM #136120).526,527 Multiple mutations have been reported.524,525,526,527,528
Clinically, cholesterol storage occurs in many tissues. Patients may develop anemia and renal insufficiency. Diagnosis is by renal biopsy, enzyme assay, or identification of the mutation.
Apolipoprotein A1 deficiency (OMIM *107680)
The gene for apolipoprotein A1 (ApoAI), on chromosome 11q23, encodes the most abundant apoprotein in high-density lipoproteins (HDL) and is a cofactor for LCAT; it also promotes efflux of cholesterol from cells. Some mutations of the gene result in defective LCAT activation525,528 and also may cause “fish-eye” disease.526,529 The numbers of reported cases are very small, and the pattern is autosomal recessive.
Individuals with familial hypoalphalipoproteinemia (HDL deficiency) have low HDL-cholesterol (HDL-C) and are at higher risk for coronary heart disease; a heterozygous mutation in ApoA1 may be evident. In the Japanese population, 6% of children have mutations of ApoA1.530
Individuals with homozygous mutations may have xanthelasma.531
Fish-eye disease (OMIM #136120)
Fish-eye disease (FED) was originally described in the late 1970s in a family with either autosomal dominant or pseudo-dominant inheritance.532,533 Homozygous mutations (autosomal recessive) of LCAT526 and ApoA1525 have been described in affected individuals.
Clinically, cardiac disease may be evident in affected individuals.
The ocular features are prominent with significant corneal opacification early in life.532
ATP-binding cassette, subfamily A, member 1 (OMIM +600046)
The ATP-binding cassette, subfamily A, member 1 (ABCA1) enzyme, the gene (ABCA1) for which is on chromosome 9q22-q31, functions in the cholesterol lipid pathway removal. Mutations in the ATP-binding cassette (ABC) family cause adrenoleukodystrophy (see entry), Zellweger syndrome (see entry), Stargardt disease,534 autosomal recessive retinitis pigmentosa,535,536 and cone-rod dystrophy.536
Deficiency of the ABCA1 also causes hypoalphalipoproteinemia or high-density lipoprotein deficiency (OMIM #604091) and Tangier disease (OMIM #205400), a rare autosomal recessive disorder found on Tangier Island off the state of Virginia. Autosomal dominant variants (hypoalphalipoproteinemia or high-density lipoprotein deficiency) may cause a mild disease or no abnormalities. Mutations in the ACADA1 cause Tangier disease and may cause familial high-density lipoprotein deficiency.537,538,539
Clinically, individuals with Tangier disease have enlarged yellow tonsils, hepatosplenomegaly, and a peripheral neuropathy; premature coronary artery disease is a feature.540,541,542
Bile Acid Metabolism
Cerebrotendinous xanthomatosis (27-hydroxylase deficiency) (OMIM #213700)
Cholestanol lipidoses (cerebrotendinous xanthomatosis) is an autosomal recessive disorder caused by mutations in the sterol 27-hydroxylase (CYP27A1) gene (OMIM #606530)545 on chromosome 2q33-qter. The protein product, sterol 27-hydroxylase, a mitochondrial cytochrome P-450 protein, catalyzes several oxidation reactions in bile acid synthesis; defective function results in reduced bile acid synthesis and accumulation of bile acid precursors and increased plasma and tissue cholestanol levels.545,546,547 Deficiency of the sterol 27-hydroxylase causes serum cholesterol to be converted to cholestanol, which accumulates in brain and other tissues. Several mutations of the CYP27A1 gene have been reported.548,549
Clinically, the disease is characterized by psychiatric disturbances (including schizophrenia) and ataxia in teenage years with subsequent neurological deterioration. Spinal disease and/or chronic diarrhea may occur. The brain, primarily white matter, accumulates cholestanol, and tendon xanthomas, prominently in the Achilles tendon, accumulate cholesterol primarily with some cholestanol. The disorder is very rare and the clinical features may include ataxia, progressive mental deterioration, and neurologic degeneration.550,551 Affected individuals may also have diarrhea.552
Ocular features include proptosis, palpebral xanthelasmas, yellow lesions in the periocular skin that represent lipocytes in the dermis, juvenile cataracts and optic atrophy corneal arcus senilis, retinal arteriolar narrowing, retinal pigment epithelial clumping, hard exudates in the retina, and optic atrophy.550,551,552,553,554,555 Vitreous “flakes” resembling “cholesterol crystals” have been described. The lens opacities are described as nuclear, corticonuclear, anterior polar, and posterior subcapsular552 and develop between the first and third decades of life.553 Cholesterol and cholestanol are elevated in cataractous lenses.556 Because of differences in clinical features, a serum cholestanol level should be considered in patients with juvenile cataract of unknown etiology.557
Other Disorders of Lipid Metabolism
Mevalonate kinase deficiency (OMIM +251170)
The gene for mevalonate kinase (MVK), on chromosome 12q24, encodes an enzyme in the cholesterol biosynthetic pathway. Deficiency results in elevated mevalonate and is inherited in an autosomal recessive pattern. Multiple mutations have been reported.560,561
Clinical features are variable. All patients have recurrent crises with fever and lymphadenopathy, hepatosplenomegaly, arthralgias, and edema. In severe cases, death occurs early. Others have developmental delay, hypotonia, myopathy, and ataxia.562
Smith-Lemli-Optiz syndrome (OMIM #270400)
The Smith-Lemli-Optiz (SLO) syndrome, an autosomal recessive disorder, is caused by mutation of the sterol 7-dehydrocholesterol reductase gene (DHCR7) (OMIM *602858) on chromosome 11q12-13. This enzyme catalyzes the last step in cholesterol synthesis. This is a metabolic disorder that causes congenital malformations. It is a relatively common disorder of individuals of northern and central European background.565 Multiple mutations have been described.566
Clinically, affected individuals have a characteristic facies and most have developmental delay, microcephaly, hypotonia, and syndactyly of the toes. Males may have hypospadias.
Chondrodysplasia punctata, X-linked dominant (OMIM #302960)
An X-linked dominant form of chondrodysplasia punctata is caused by deficiency of the emopamil-binding protein involved in cholesterol biosyntheses; the gene (EBP) (OMIM *300205) is on chromosome Xp11.23-p11.22; multiple mutations have been described.571,572,573
In addition to epiphyseal stippling and a predominance in females, the clinical features include linear or whorled atrophic and pigmentary lesions of the skin, coarse hair and alopecia, cataracts, and short stature with rhizomelic shortening of the limbs and craniofacial defects.572,574
Disorders of Fatty Acid Transport and Mitochondrial Oxidation
Trifunctional protein deficiency (OMIM #609015)
The trifunctional protein catalyzes three steps in mitochondrial oxidation of fatty acids and includes the long-chain 3-hydroxyacly-CoA dehydrogenase (LCHAD) (OMIM #609016). The protein is a heterocomplex composed of both alpha and beta subunits. The autosomal recessive disorder LCHAD (OMIM #60916) is caused by mutations of hydroxyacyl-CoA dehydrogenase/3-detoacyl-CoA thiolase/enoyl-CoA gene, alpha subunit (HADHA) (OMIM *600890)576,577 on chromosome 2p23 or the beta subunit, hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enlyl-CoA hydratase (HADHB) (OMIM *143450), on chromosome 2p23, causes trifunctional protein deficiency.578 Multiple mutations have been described.577,579,580
Clinically, patients have early and profound psychomotor retardation and die early in infancy. Mutations of the HADHA gene (LCHAD) results in sudden infant death syndrome, hypoglycemia, and cardiomyopathy581,582 Mutations of the HADHB gene cause three different clinical forms: a severe neonatal onset with cardiomyopathy and early death; a hepatic form; and later onset form with skeletal myopathy and myoglobinuria.583
Organic Acidemias and Disorders of Fatty Acid Oxidation
Organic Acidemias
Biotinidase deficiency (OMIM #253260)
Biotinidase catalyzes the hydrolysis of biocytin and deficiency causes a late-onset multiple carboxylase deficiency because of the inability to reutilize biotin. The disease is inherited in an autosomal recessive pattern. Although multiple mutations of the gene (BTD) (OMIM *609019) on chromosome 3p25 have been reported,588 some are more common.589
Clinical features include alopecia and seborrheic dermatitis; rarely, infants may develop infantile spasms (severe seizures in infancy). Some individuals with significant reduction in biotinidase activity are normal.590
Propionic acidemia (OMIM #606054)
A deficiency of propionyl-CoA carboxylase causes propionic acidemia and is inherited as an autosomal recessive disorder. Genes encoding the enzyme include an alpha (PCCA) (OMIM *232000) subunit, on chromosome 13q32, and a beta subunit (PCCB) (OMIM *232050), on chromosome 3q21-q22. The enzyme catalyzes propionyl-CoA and mutations of either subunit may cause disease; multiple mutations for each have been identified.593
Clinical features include developmental delay with intermittent vomiting, intolerance to protein, neutropenia, and intermittent thrombocytopenia. Affected individuals may have an acute encephalopathy and/or cerebral palsy.
Reported ocular features include optic atrophy.594
Glutaric acidemia type 1 (OMIM #231671)
Glutaric acidemia type 1 is an autosomal recessive disorder caused by deficiency of the enzyme glutaryl-CoA dehydrogenase. Multiple mutations of the gene (GCDH) (OMIM *608801), on chromosome 19p13.2, have been identified.595
Clinically, a movement disorder begins relatively early in life with symptoms of basal ganglia disease.596 Manifestations can vary.597
Vitamin D Metabolism
Inherited Porphyrias
Congenital erythropoietic porphyria (OMIM #263700)
Congenital erythropoietic porphyria (Gunther disease), inherited in an autosomal recessive pattern, is caused by deficiency of uroporphyrinogen III synthase (URO-synthase); the gene (UROS) (OMIM *606938) is on chromosome 10q25.2-q26.3. Some enzymatic activity is needed for production of red blood cells.604 Affected individuals have an overproduction of series I porphyrins. Many mutations have been identified.605,606
Clinically, affected individuals are sensitive to light, which causes severe blistering and scarring of the skin; hypo- and hyperpigmentation develop. The phenotype is related to URO-synthase activity, and affected individuals experience hemolysis. Although onset is usually in childhood, the age varies.
Copper Metabolism
Menkes disease (OMIM #309400)
Menkes disease613 is an X-linked recessive disorder caused by defective cellular copper export614,615; copper accumulates intracellularly in some tissues (intestinal mucosa and kidney) and is deficient in others. The disease is caused by deficiency of the copper-transporting P-type ATPase, ATP7A, the gene for which (MNK)616,617,618 (OMIM *300011) is on chromosome Xq12-q13. The protein is in the trans-Golgi network.619 The gene has approximately 55% homology (similar sequence) to ATPase, Cu(2+)-transporting, beta polypeptide (ATP7B), the protein responsible for Wilson disease. Multiple mutations and deletions have been described620,621,622,623 including a milder form.620 The occipital horn syndrome (X-linked cutis laxa) is caused by mutations of the same gene.624,625
Menkes disease is usually lethal relatively early in life and presents early with significant neurologic impairment.626,627 Facial features and distinctive hair (“kinky” and lightly pigmented) develop. Myoclonic seizures and progressive deterioration lead to death in early childhood. Radiological features may useful diagnostically. There is a spectrum of severity.628 A mild form, X-linked cutis laxa (occipital horn syndrome) (OMIM #304150), was categorized initially as a connective tissue disease with mild neurologic impairment.620 Mild forms may exist in heterozygotes females.629 Serum copper and ceruloplasmin are decreased after approximately 2 weeks of age.
Wilson disease (OMIM #277900)
Wilson disease is an autosomal recessive disorder caused by deficiency of ATPase, Cu(2+)-transporting, beta polypeptide; the gene (ATP7B) (OMIM *606882) is on chromosome 13q14.3-q21.1. Two proteins are formed and one is localized to the Golgi apparatus and one to the cytosol.635 The manifestations are related to copper toxicity with impaired intracellular transport of copper into hepatocytes and incorporation into ceruloplasmin. Copper accumulates in the liver, kidney, and basal ganglia; elevated plasma copper levels result in widespread toxicity. Many mutations have been described and some reflect a founder effect.636,637,638,639,640,641,642,643,644,645,646,647,648
Clinically, the range of age-of-onset is wide varying from early childhood to over 50 years of age, and the variability is attributed to the large number of mutations.649,650 Presentation may occur as fulminate liver failure, psychiatric disease, motor dysfunction, or Fanconi syndrome. In most affected individuals, copper and ceruloplasmin levels are decreased.650
Ocular features include a brown to green discoloration in the peripheral cornea at the level of Descemet membrane (Kayser-Fleiser ring)651 (Fig. 14a) and rarely may be unilateral.652 Although common in patients with neurologic degeneration or psychiatric symptoms, they may be absent in patients with liver disease.653 Copper deposition at the level of the anterior capsule of the lens (“sunflower cataract”) is also characteristic of the disease654 (Figs. 14b and 15); rarely the vision is decreased.655 The Kayser-Fleiser ring may be the initial manifestation656,657,658 and conversely, patients with systemic manifestations may not have corneal deposits.659 Reversible electroretinographic abnormalities have been reported.660 The corneal deposits may resolve with zinc and/or penicillamine treatment.661
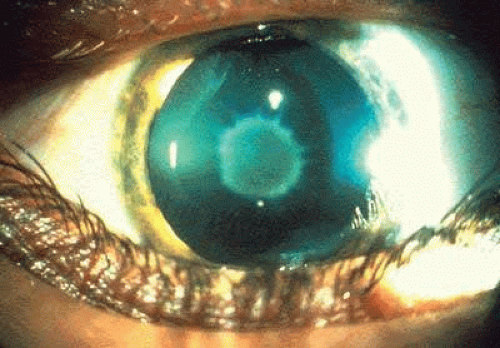 Fig. 14. Wilson disease. Cornea and lens of patient with Wilson disease. a: The Kayser-Fleisher ring and central anterior capsular cataract is copper deposition. b: The copper deposition in the cornea is at the level of descemet membrane and the cataract at the level of the anterior capsule is faintly visible. |
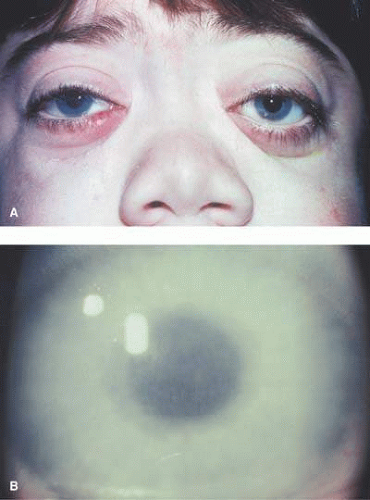 Fig. 15. Wilson disease. The cataract at the level of the anterior capsule has a bluish coloration. (Courtesy of Irene H. Maumenee MD) |
Disorders of Iron Metabolism
Hereditary hyperferritinemia-cataract syndrome (OMIM #600886)
Hereditary hyperferritinemia-cataract syndrome is an autosomal dominant disorder caused by elevated levels of the light chain of ferritin. The gene (FTL) (OMIM *134790) is located on chromosome 19q13.3-q13.4 and the diagnosis is made on the basis of elevated serum levels of the light chain of ferritin662 or by mutational analysis. Ferritin is found in all cells and its function is iron concentration and storage; it protects the cell against iron. Mutations in the iron-responsive element (IRE) in the 5-prime noncoding region of the ferritin light chain gene cause the disease. Many mutations have been reported from diverse regions of the world.663,664,665,666,667,668,669,670,671,672,673,674,675,676,677,678,679,680 Affected individuals have normal serum iron and transferring but elevated levels of ferritin.
Systemic features are minimal, if any; minor abnormalities are evident on liver biopsy.
Pantothenate kinase-associated neurodegeneration (Hallervorden-Spatz) (OMIM #234200) (OMIM #607236)
Pantothenate kinase-associated neurodegeneration (Hallervorden-Spatz) and HARP (hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration) are allelic autosomal recessive disorders caused by deficiency of the pantothenate kinase protein. The gene (PANK2) (OMIM *607236) is located on chromosome 20p13-p12.3 and the protein product is a regulatory enzyme in CoA biosynthesis that catalyzes phosphorylation of pantothenate (vitamin B5), N-pantothenoylcysteine, and pantetheine in the cytoplasm. Multiple mutations have been identified.681,682
Clinically, most patients present with either gait or writing abnormalities; some patients have initial symptoms of psychomotor delay or tics similar to Tourette syndrome.683 Over time, most develop corticospinal signs, dysarthria, dystonia, and rigidity. Magnetic resonance imaging (MRI) alterations are characteristic and include iron deposition in the basal ganglia.684,685
Friedreich ataxia (OMIM #229300)
Friedreich ataxia is a rare autosomal recessive disorder caused by mutations of the frataxin (OMIM *606829) gene (FRDA), on chromosome 9q13; the protein product is involved in mitochondrial iron metabolism. Although some patients have point mutations, most cases are caused by a GAA-repeat expansion in intron 1.690,691
Clinically, patients have spinocerebellar ataxia and other neurologic symptoms with some correlation of the features associated with the shorter repeat allele.690 In some cases, cardiac manifestations are prominent features.692
Mucopolysaccharidoses
The mucopolysaccharidoses are lysosomal storage disorders.
Mucopolysaccharidosis I-H (Hurler syndrome) (OMIM #607014)/Mucopolysaccharidosis I-S (Scheie syndrome) (OMIM #60716)/Mucopolysaccharidosis I-HS (Hurler-Scheie syndrome) (OMIM #60715)
Mucopolysaccharidosis I-H (Hurler syndrome), mucopolysaccharidosis I-S (Scheie syndrome), and mucopolysaccharidosis I-HS (Hurler-Scheie syndrome) are autosomal recessive diseases caused by mutations of the gene IDUA (OMIM *252800), on chromosome 4q16.3. The gene encodes α-L-iduronidase, a lysosomal enzyme that hydrolyzes the terminal α-L-iduronic acid residues from glycosaminoglycans dermatan sulfate and of heparan sulfate. The Scheie syndrome, a milder disease, is allelic and intermediate forms are termed Hurler-Scheie. Multiple mutations have been reported698,699 with the milder forms usually being due to missense mutations.700
Hurler infants are usually normal at birth and develop signs and symptoms in the first year of life. Clinical features of the Hurler syndrome include coarse facies, mental retardation, hernias, boney disease, and hepatosplenomegaly. The mean age of diagnosis is 9 months.701 Individuals with the Scheie syndrome have stiff joints and aortic regurgitation; they have normal intelligence and survive until late in life.702 Diagnosis is usually in teenage years but many not occur until adulthood. The intermediary disorder, Hurler-Scheie, exhibits a wide range of signs and symptoms between the two more extreme phenotypes.
Ocular features of all forms of the disorder include corneal clouding, retinal degeneration, and optic nerve head swelling with papilledema.703,704,705,706,707,708 In Hurler disease, corneal clouding is the most prominent and consistent feature,708 with rare exceptions.709 Glaucoma may occur.710 In Hurler-Scheie, corneal clouding and retinal degeneration are consistent features; glaucoma may occur.707,708,711,712 In Scheie syndrome, corneal clouding and retinal degeneration occur; angle closure glaucoma also may develop.707,708,713
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


