Choanal stenosis and atresia
Congenital nasal piriform aperture stenosis
Midnasal stenosis
Nasal masses
Dermoid cyst
Encephalocele
Glioma
Hemangioma
Nasolacrimal duct cyst
Deviated nasal septum
Rhinitis of the newborn
Congenital Nasal Piriform Aperture Stenosis
The piriform aperture is the most anterior aspect of the bony nasal airway. It has a pear shape and anatomically distinct bones contribute to the aperture. The horizontal processes of the maxilla contribute inferiorly. The frontal processes of the maxilla (inferiorly) and nasal bones (superiorly) constitute the lateral aspect of the aperture. The aperture is divided in the midline by the anterior nasal spine and septum. CNPAS is defined as horizontal narrowing of the lateral walls resulting in a smaller aperture. The narrowing is typically measured at the level of the inferior meatus.
Etiology
Embryologically, the piriform aperture is derived from fusion of the medial nasal prominences (floor of the aperture) and fusion of the lateral nasal prominences and maxillary prominences (lateral bones of aperture). Therefore, it has been hypothesized that CNPAS can be caused by either a deficiency in the primary palate (floor of the aperture) or bony overgrowth/dysplasia of the nasal processes of the maxilla. Both etiologies would result in horizontal narrowing of the lateral walls of the piriform aperture.
Epidemiology
CNPAS is an uncommon cause of congenital nasal airway obstruction and the true incidence is unknown.
Pathogenesis
The embryological development of the nose begins early in the fourth week of gestation [4]. The nose is derived from the single median frontonasal prominence and the paired maxillary prominences. The prominences arise from the proliferation and migration of neural crest cells during the fourth week.
The frontonasal prominence gives rise to bilateral nasal placodes by the end of the fourth week. The mesenchyme at the periphery of the placodes proliferates creating medial and lateral nasal prominences and central depressions called nasal pits. The nasal pits are the primordium of the nostrils and nasal cavities.
With medial migration of the maxillary prominences, the medial nasal prominences begin to fuse and form the primary palate (Fig. 1a, b). Concurrent fusion of the lateral nasal prominences with the maxillary prominences forms the lateral bones of the piriform aperture.
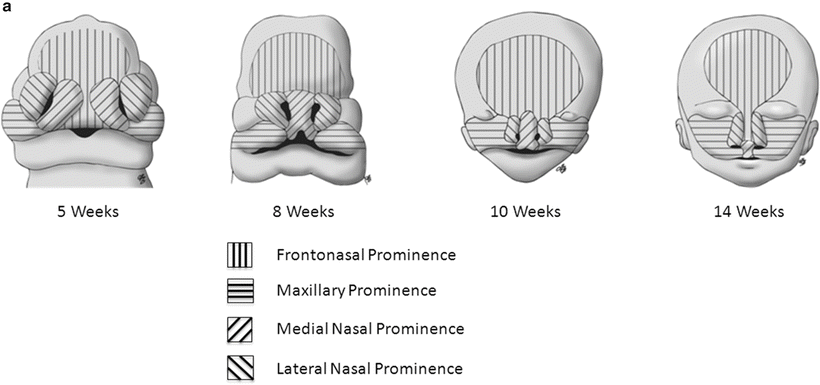
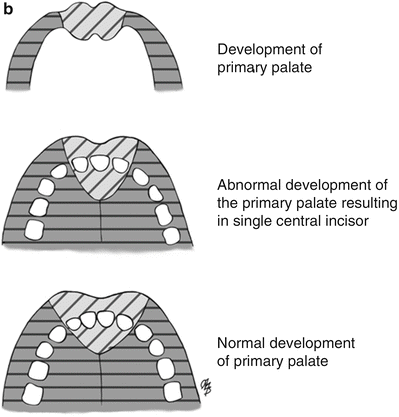
Fig. 1
(a, b) Drawings illustrating the embryological development of the piriform aperture and primary palate
A deficiency in the bulk of the primary palate primordium may result in CNPAS by allowing medial migration of the palatal shelves during the 6th week of gestation that is closer to the midline than usual. The deficient primary palate would account for the narrow aperture, triangular shaped palate, and single central incisor associated with CNPAS (Fig. 2). The resultant “over-migration” of the palatal shelves may account for the observed ridge of bone along the inferior aspect of the palate.
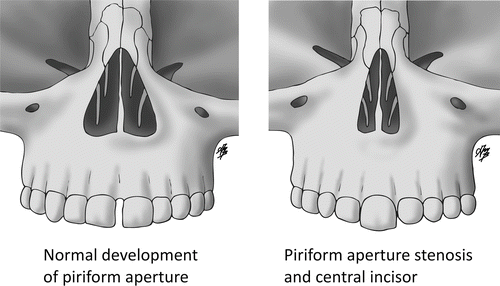
Fig. 2
Drawing illustrating the normal and abnormal size and shape of the piriform aperture
The other possible cause of CNPAS is the overgrowth of the medial maxillary bones. Imaging studies frequently show thickened bone in this region.
Clinical Presentation
The clinical presentation of CNPAS is similar to the presentation of any condition that causes nasal obstruction. Typical signs and symptoms include stertor, tachypnea, obstructive apnea, feeding difficulty, respiratory distress, and cyclical and paradoxical cyanosis. Crying usually relieves the respiratory symptoms as the infant shifts their breathing from nasal to oral routes.
On physical examination, the nasal inlet is visibly narrow and there may be difficulty passing a suction catheter or small endoscope. There may be other craniofacial abnormalities noted during the routine head and neck examination such as a triangular shaped palate, a bony ridge under the palate, and other maxillary abnormalities.
Diagnosis
Although the suspicion for CNPAS is initiated by the clinical presentation and physical examination, the diagnosis is typically confirmed with computed tomography scanning of the craniofacial skeleton. The width of the piriform aperture is less than 11 mm [1], there may be bony thickening of the medial maxilla and there may be a single central incisor tooth bud (Figs. 3 and 4). The presence of a central incisor is pathognomonic for CNPAS and requires further investigations to rule out other midline brain anomalies.
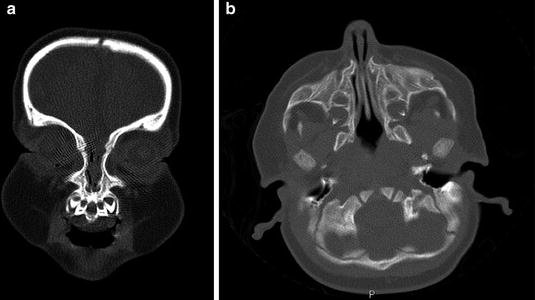
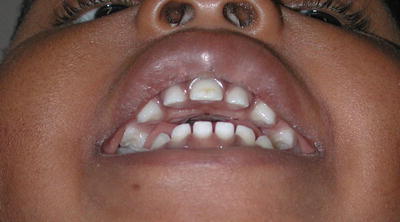
Fig. 3
Computed tomography imaging of a child with congenital nasal piriform aperture stenosis. (a) Coronal image demonstrating the central incisor tooth bud. (b) Axial image demonstrating the narrow aperture and prominence of the maxillary bone
Fig. 4
Clinical photograph of an older child with a central incisor
Management
The management of CNPAS primarily depends on the severity of the respiratory symptoms of the patient. Management options include observation, nasal hygiene, topical steroids and decongestants, serial nasal dilatation, nasal stenting, and surgical correction. The medical literature contains several small case series describing all of the management options listed above.
The larger case series by Van Den Abbeele et al. [3] and Visvanathan and Wynn [2] confirm that conservative or surgical intervention depends on the severity of symptoms. Both studies recommend an initial conservative approach with nasal steroids and topical decongestants for 2 weeks before considering surgery. Their criteria for surgical intervention included sleep apnea, failure to thrive, and persistent dependency on airway assistance at the end of the 2-week trial.
The most common surgical approach employed in the correction of CNPAS is a sublabial incision and mucosal elevation to reveal the piriform aperture. A drill is then used to expand the aperture inferiorly and laterally. Care is taken to avoid injury to the nasolacrimal duct laterally and tooth buds inferiorly. Postoperative nasal stenting for several weeks is used to prevent restenosis.
Multidisciplinary Considerations
CNPAS may occur in isolation or as part of the holoprosencephaly spectrum. The suspicion for other central anomalies is increased if there is a single central incisor (Figs. 2 and 3). Specifically, patients require an endocrinological assessment for pituitary hormone deficiency, an MRI of the brain to assess the corpus callosum and pituitary gland, and a genetics consultation if there are other congenital anomalies. Patients may also require dental and orthodontic consultation following the eruption of the secondary dentition.
Choanal Atresia
Choanal atresia is the end result of a failure to establish communication between the nasal cavity and nasopharynx during embryological development. In essence, an atretic plate remains that obstructs the region of one or both choanae. The choanae are bounded by the sphenoid superiorly, the medial pterygoid plate laterally, the vomer medially and the horizontal portion of the palate inferiorly. The choanae may be stenosed or completely blocked (atresia) by bone and membranous tissue (70 % of cases) or by bone only (30 % of cases). In addition to the atretic plate of bone and/or membranous tissue, medialization of the medial pterygoid plate and thickening of the posterior vomer often contribute to the stenosis and atresia.
Etiology
Many theories have been proposed to explain the development of choanal atresia. The four most popular theories include: (1) persistence of the bucconasal membrane of Hochstetter; (2) misdirection of neural crest cell migration; (3) persistence of the buccopharyngeal membrane from the foregut; and (4) abnormal persistence or location of mesodermal adhesions in the naso-choanal region [6].
Molecular models have also been linked to the development of choanal atresia. For example, it has been shown that mice with a retinaldehyde dehydrogenase 3 (Raldh3) knockout mutation develop choanal atresia. This work suggests that retinoic acid metabolism is linked to fibroblast growth factor (FGF) expression and eventual perforation of the nasobuccal membrane. Another example is the observed increased incidence of non-syndromic choanal atresia in the children of mothers treated for hyperthyroidism with thionamides. Subsequent animal studies have demonstrated that elevation of thyrotropin alters the expression of FGF, FGF receptors which may play a role in the development of choanal atresia.
Choanal atresia is a hallmark feature of CHARGE syndrome. A mutation in the chromodomain helicase DNA-binding protein 7 (Chd7), located on chromosome 8q12.1, is identified in 60–80 % of children with CHARGE syndrome. Chd7 regulates other genes that control cell-lineage specification and rRNA biogenesis in the nucleolus. The pathogenic mechanism on organ systems such as choanal development is not fully understood.
Epidemiology
Choanal atresia is the most common congenital nasal anomaly with an estimated incidence of 1 in 5,000–8,000 live births. Choanal atresia is more likely to occur in females and be unilateral. When unilateral, the right choana is twice as likely to be affected. Choanal atresia can occur in isolation or be associated with several other conditions such as CHARGE, Apert, Crouzon, Pfeiffer, Antley–Bixler, Marshall–Smith, Schinzel–Giedion, and Treacher Collins syndromes. Approximately 30 % of children with choanal atresia have CHARGE syndrome based on clinical criteria.
Pathogenesis
During the fourth week of gestation, the nasal placodes become depressed and form nasal pits. From the fourth to sixth week, the nasal pits deepen to form primitive nasal sacs. The nasal sacs grow dorsally and remain separate from the oral cavity by the bucconasal membrane of Hochstetter (Fig. 5). By 7 weeks, the bucconasal membrane ruptures allowing communication of the oral and nasal cavities. The resultant primitive choanae are located just posterior to the primary palate. As the secondary palate develops by fusion of the lateral palatine processes, the choanae assume a more posterior location [4].
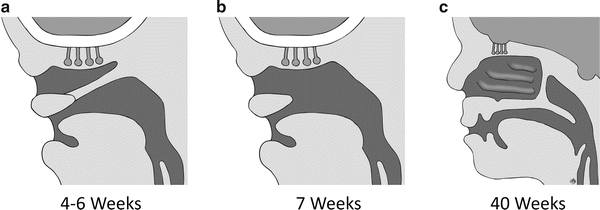
Fig. 5
Drawings illustrating the normal and failed embryological development of the nasal choanae. (a) The nasal and oral cavities are separated by the bucconasal membrane. (b) The normal rupture of the bucconasal membrane. (c) Persistence of the bucconasal membrane at birth
The pathogenesis of choanal atresia is poorly understood. However, the description of the embryological development provided above renders the theory of failure to disrupt the bucconasal membrane of Hoschstetter plausible (Fig. 5). Further elucidation of the molecular and genetic theories may help to explain the mechanism by which the bucconasal membrane fails to rupture.
Clinical Presentation
The clinical presentation of choanal stenosis and atresia is consistent with the presentation of any condition that causes nasal obstruction: stertor, obstructive apnea, cyclical cyanosis, etc. Newborns with bilateral choanal atresia present with immediate respiratory distress and require prompt securing of the airway with intubation. The suspicion for choanal atresia in these instances is reinforced by the inability to pass a suction catheter into the nasopharynx. Children with unilateral choanal atresia are often diagnosed later in life as they present with complaints of persistent unilateral nasal obstruction and rhinorrhea [5].
Infants and children with isolated choanal atresia typically have a normal physical examination (excluding the signs and symptoms of nasal obstruction). However, if the child presents with other anomalies, the clinician should determine if the choanal atresia is part of a genetic syndrome. The most commonly associated condition is CHARGE syndrome (Coloboma of the eye, Heart malformations, Atresia of choanae, Retardation in growth and development, Genital hypoplasia, Ear anomalies).
Diagnosis
Choanal atresia can be diagnosed with a thorough clinical examination of the nose. The failure to observe fogging of a cold metal speculum placed in front of the nostrils, the movement of a wisp of cotton wool, or the inability to auscultate air flow with the bell of a stethoscope suggests a blocked nasal passage. The visualization of a blind ended nasal cavity with endoscopy confirms the diagnosis of choanal atresia.
The diagnosis is further confirmed with computed tomography scanning of the nose, paranasal sinuses, and skull base (Fig. 6). The images are used to characterize the location and nature of the atresia (bony versus bony and membranous). The images may also demonstrate thickening of the posterior vomer and medialization of the medial pterygoid plate.
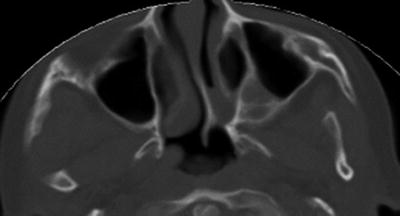
Fig. 6




Computed tomography image (axial view) of a child with unilateral (left) choanal atresia. The image demonstrates a combined bony and membranous atretic plate, deviated vomer, and medialized left medial pterygoid plate
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
