Fig. 1
Crouzon & Apert Syndromes. (a) Front, & (b) Side view of Crouzon Syndrome. Note brachycephaly (short skull in AP dimension, with flat occiput), frontal bossing, maxillary hypoplasia with mandibular prognathism, shallow orbits and hypertelorism. Similar features are seen in Apert syndrome (c—Front, d Side), although proptosis is more evident in this infant because his eyes are open. Syndactyly of all five fingers (“mitten hand” deformity; not shown here) in Apert syndrome would easily distinguish these otherwise very similar-appearing conditions. Courtesy of CHOP Craniofacial Program, Permission Obtained
A syndrome refers to a recurrent pattern of malformation arising from a single underlying etiology. A well-known example is Trisomy 21 or Down syndrome. The multiple associated features affect various organ systems ranging from the characteristic facies to major structural defects, including cardiac septal defects, Tetralogy of Fallot, duodenal atresia, and anal stenosis. The findings are collectively identified as a syndrome because of the single underlying cause, an extra 21st chromosome. In contrast, when a collection of anomalies is recognized to occur together more frequently than expected by chance, but when no unifying etiology can be established, it is referred to as an association. Many associations, such as VACTERL or OAVS (Oculo-Auriculo-Vertebral Spectrum), exhibit significant and even life-threatening airway anomalies. Finally, a pattern of anomalies that results from a single primary anomaly or mechanical force with secondary effects on other structures is referred to as a sequence. Pierre-Robin sequence, in which primary micrognathia results in glossoptosis and cleft palate, is an example of a sequence with important airway implications.
Among the airway anomalies with an identifiable genetic etiology, the nature of the genetic lesions leading to these disorders is equally diverse, ranging from the involvement of an entire chromosome (e.g., Trisomy 18 or 21), to a single base pair change in a critical gene, as with the FGFR2 mutation in Crouzon syndrome. Overall, it is estimated that 10 % of all birth defects are due to a chromosomal abnormality [2]. Aneuploidy refers to the presence of an abnormal number of chromosomes and includes the important syndromes such as Trisomy 18 and 21, both of which have been associated with significant airway anomalies (Table 1). Deletions or duplication of genetic material can also affect just a portion of a chromosome and also cause significant life-threatening airway malformations.
Table 1
Airway anomalies associated with Trisomy 13, 18, and 21
Trisomy 18 | Cleft lip/palate |
Tracheoesophageal fistula | |
Pulmonary hypoplasia or aplasia | |
Trisomy 21 | Laryngeal web |
Laryngeal cleft | |
Subglottic stenosis | |
Tracheal stenosis | |
Tracheoesophageal fistula |
For nearly half of all congenital malformations, a comprehensive diagnostic evaluation will fail to reveal an underlying genetic etiology [3]. This may be due to the limitations of available diagnostic techniques and also likely reflects our incomplete understanding of human genetics. The failure to identify an associated genetic change also highlights the fact that a variety of nongenetic factors can contribute to human birth defects. Environmental factors, including infectious agents, maternal disease states, drugs, chemicals, and radiation, have all been linked to disrupted human development. While beyond the scope of this review, exposures to agents like diphenylhydantoin (Dilantin) or vitamin A (e.g., excessive supplemental vitamin intake, isotretinoin) can lead to altered craniofacial development and cleft lip/palate, while other drugs including warfarin can lead to choanal stenosis. Our understanding of how subtle environmental exposures, including dietary factors or common environmental contaminants interact with genetic factors to contribute to the formation of congenital malformations, is rudimentary, but future research in this area will likely greatly enhance our understanding of many complex congenital airway disorders.
Similarly, recent research has begun to elucidate how chemical modifications of DNA or its supporting structure (chromatin) that leaves the DNA sequence itself unchanged, can alter normal development and result in human disease. These epigenetic changes can even be passed on from one generation to the next and often reflect a response to environmental cues such as starvation and obesity. Examples exist of known human disorders in which the normal epigenetic marks are disrupted. These include Prader-Willi and Angelman syndromes in chromosome 15q11, and Beckwith-Wiedemann syndrome in chromosome 11p15. It is possible that epigenetic changes may contribute to complex and likely multifactorial airway malformations. For example, although laryngotracheal clefts and tracheosophageal fistulas have been associated with several syndromes (Tables 2 and 3), the precise etiology of these complex disorders remain almost completely unexplored, and such areas of investigation will also likely contribute greatly to our understanding of the etiology of these disorders over the coming years.
Table 2
Syndromes associated with TEF/EA
Disorder | Key features | Etiology/Gene |
|---|---|---|
22q11.2 microdeletion syndrome | Congenital heart defects, hypocalcemia, immune deficiency (thymic aplasia), characteristic facial features | Deletion of chromosome 22q11.2 |
Trisomy 18 | Growth retardation, microcephaly, microphthalmia, malformed ears, microretrognathia, clenched fists, rocker-bottom feet, congenital heart disease | Trisomy 18 |
Trisomy 21 | Characteristic facial features, infantile hypotonia, intellectual disability, congenital heart disease, duodenal atresia, hypothyroidism | Trisomy 21 |
CHARGE | Coloboma of the eye, Heart defects, choanal Atresia, Retardation of growth/development, GU anomalies, Ear anomalies | CHD7 gene (AD) |
Feingold | Microcephaly, limb malformations, esophageal/duodenal atresia, hand/foot malformations, cardiac, renal, vertebral malformations, intellectual disability | MYCN gene (AD) |
OAVS | Facial asymmetry with malar, maxillary, and mandibular hypoplasia, microtia, ear tags, ocular anomalies (e.g., epibulbar dermoids), and vertebral anomalies | Unknown |
Optiz-G BBB | Hypertelorism, broad nasal bridge, anteverted nares, grooved nasal tip, cleft lip/palate, congenital heart disease, imperforate anus, hypospadias, widow’s peak, agenesis of corpus callosum | MID1 (X-linked) |
VACTERL | Vertebral anomalies, Anal atresia, Cardiac defects, TEF/EA, Renal, Limb anomalies | Unknown |
Table 3
Syndromes associated with laryngeal clefts/LET clefts
•$Opitz-G BBB syndrome |
•$Pallister-Hall syndrome |
•$Rhizomelic chondrodysplasia punctata syndrome |
•$Trisomy 21 |
Comprehensive Diagnostic Approach to the Infant with An Airway Disorder
Physical Assessment
The genetics evaluation of a neonate with airway malformation is guided by the type and location of the malformation, a specific medical history and a detailed physical examination. History: A complete prenatal history should be obtained, including results of any prenatal testing. Maternal and paternal ages at conception are important to discern, since there is an increased risk of chromosomal nondisjunction and subsequent chromosomal anomalies that occurs with increasing maternal age at conception. Similarly, advanced paternal age is associated with increasing risk for single gene disorders, such as achondroplasia. The use of assisted reproductive techniques to achieve pregnancy and use of donor gametes should be identified. In addition, the results of any imaging studies such as ultrasound evaluation, fetal echocardiogram and fetal MRI in cases where such modalities have been utilized should be reviewed. For example, an ultrasound assessment of abnormal levels of amniotic fluid may provide a clue to an underlying malformation and potential diagnosis. Oligohydramnios or decreased amniotic fluid could indicate leakage of fluid or a malformation of the renal or urinary tract structures. Instead, excess fluid, or polyhydramnios may indicate poor fetal swallowing and relate to a neurologic condition in the fetus precluding normal movement or a gastrointestinal malformation such as tracheoesophageal fistula. Anatomic differences of the uterus can result in fetal constraint and a deformational effect on airway structures. Such underlying issues may have already been identified on imaging studies of the pregnancy.
A general maternal health history such as the presence of diabetes, hypertension, drug and alcohol exposures, infections, and use of medications is important to elicit. Teratogenic exposures, to ethanol for example, may cause structural effects on the exposed fetus. There are numerous known teratogens, many with specific associated structural anomalies and dysmorphisms. These include well-documented human teratogens such as thalidomide, warfarin, valproate, hydantoin, and trimethadione. Some can display a pronounced effect on airway structures. For example, fetal warfarin exposure can result in severe nasal hypoplasia, compromising the upper airway. Similarly, oral isotretinoin use, (a synthetic retinoic acid derivative used for dermatologic disorders), particularly early in pregnancy, can cause severe craniofacial malformations. The effect of a given teratogen may vary depending on the timing, dosage and duration of the exposure. In general, exposures early in gestation and at relatively higher doses have more pronounced effects on fetal development. Ascertainment of teratogen exposure is crucial, since many of these effects can mimic a chromosomal disorder; however the adjunct genetic testing and recurrence risk counseling would be quite different.
In evaluating the infant, documenting data on fetal activity, fetal size, position, evidence of fetal distress and gestational age is highly relevant to the differential diagnosis. For example, a case of a neonate with hypotonia at birth who has a history of poor fetal movement and breech presentation suggests a long-standing developmental issue rather than simply perinatal depression.
In many pregnancies, genetic studies including screening analyses such as standard maternal serum screening and more recently, noninvasive prenatal testing (NIPT) may have been performed. This latter screening test utilizes cell-free DNA circulating in the maternal peripheral blood and sequencing-based techniques to assess for commonly encountered aneuploidies such as Trisomy 21. Data from genetic diagnostic tests such as chorionic villus sampling (CVS) or amniocentesis where karyotyping and/or chromosomal microarray may have been performed should also be reviewed.
It is important to document a complete three-generation family history. Eliciting a history of consanguinity in a couple increases the likelihood of a recessive condition in the infant. Consideration of seemingly unrelated birth defects, such as cardiac or renal anomalies in other family members may provide a clue to an underlying diagnosis in the proband. Intellectual disability in close relatives is also important to document. In addition, a history of multiple (>2) unexplained miscarriages may be suggestive of an underlying chromosomal predisposition such as the presence of a balanced translocation carried by one of the parents, that may be related to the proband’s condition. Data from a formal pedigree analysis can be quite helpful in ascertaining an underlying pattern of inheritance, such as an X-linked recessive disease with affected males, relatively unaffected females, and transmission on the maternal side of the family.
Dysmorphology Exam
This detailed physical exam assesses for defects in the structure or morphology of an organ or organ system that reflects an abnormal developmental process. In assessing for congenital defects and in order to develop an appropriate differential diagnosis, a detailed physical exam with emphasis on the craniofacial exam is performed and measurements are compared to published age-matched standards [4]. The initial assessment begins with the newborn growth parameters, adjusted for any prematurity. The height, weight, and head circumference will provide a snapshot view of fetal health and intrauterine environment. For example, head-sparing intrauterine growth retardation where HC >length >weight may reflect poor nutrition or placental insufficiency, while microcephaly might instead suggest underlying neurological malformation with a genetic etiology or infectious agent such as cytomegalovirus. The dysmorphology exam focuses on assessing the patient for anatomic features that diverge from normal standards with the understanding that some might be familial variants. The shape and size of the head and fontanelles and assessment of the cranial sutures should be obtained. The presence of craniosynostosis may significantly narrow down the diagnoses under consideration and an unusual head shape or size would prompt imaging studies that might reveal a brain malformation (Fig. 2a, b). In further assessing the head, notation of any scalp defects, differences in the shape of the forehead, eyebrows, and distribution of hair should be sought. The spacing of the eyes with measurements of the canthi and palpebral fissure lengths is performed. The presence of colobomata, cataracts, and the surrounding structures such as the presence of epicanthal folds or downward or upward slant of the palpebral fissures is also documented as these can be characteristic findings of some syndromes. Careful examination of the ears, assessing their placement, rotation and appropriate folding of the ear is important as is assessment for auricular pits and tags which can be specific for certain disorders. The nose is evaluated for proper formation of the bridge, alar nasi, nares, and nasal tip. Patency of the chonae is also assessed. The mouth and throat are evaluated for evidence of defects or clefts of the lip and/or palate. The shape of the palate and the uvula are examined, with direct implications for functional issues of the airway. The mouth is carefully examined for findings such as natal teeth, gum abnormalities, lip pits, frenulae, and tongue malformations that might be suggestive of a diagnosis. Evaluation of the chin is critical in a patient with airway involvement and micrognathia and retrognathia can be part of distinct syndromic entities. They can also occur as isolated malformations. Finally, the neck is evaluated for webbing, excess nuchal skin, or bony cervical abnormalities, which would prompt a subsequent cervical spine stability evaluation, affecting airway management.
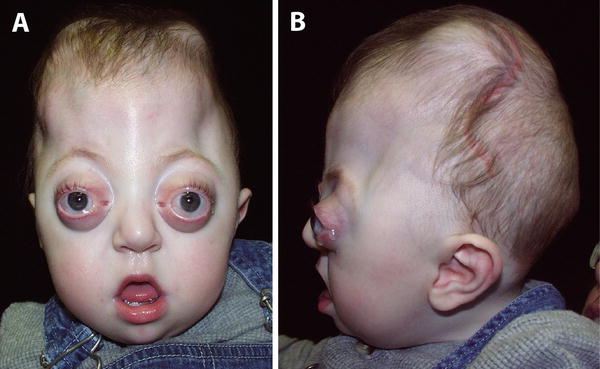
Fig. 2
(a, b)—Pfeiffer Syndrome. (a)—Front, (b)—Side. Note the short, tall skull (turribrachycephaly), maxillary hypoplasia with mandibular prognathism, shallow orbits with striking ocular proptosis, small nose, flat nasal bridge, and irregular skull shape due to the presence of craniosynostosis. The presence of broad thumbs and toes (not shown) is also characteristic of this disorder. Courtesy of CHOP Craniofacial Program, Permission Obtained
Examination of the chest and thorax notes any bony deformities such as pectus or narrow chest and internipple distances. Abnormalities should prompt imaging studies such as chest X-ray evaluating for bone malformations, while presence of a murmur would prompt cardiac evaluation. The abdominal exam assesses primarily for evidence of abdominal wall defects such as omphalocele and for the presence of organomegaly, which is more suggestive of an inborn error of metabolism. When examining the umbilical cord, a 2-vessel cord with only a single umbilical artery may reflect the presence of a renal anomaly. The genitourinary exam assesses external genitalia where anomalies may be associated with internal malformations of the GU tract as well. Patency of the anus and its placement are assessed as this finding may present as part of a recognizable constellation of findings in a syndrome. The extremities are carefully examined for structural integrity of the digits, their formation and placement and palmar creases. Often, limb anomalies particularly those of the hands and feet can be a vital clue to an underlying diagnosis.
A dermatologic evaluation to examine for the presence of phakomatoses or skin manifestations that signal an underlying genetic diagnosis is important. For example, multiple café au lait spots or neurofibromas can be associated with neurofibromatosis type I. The irregular swirly pigmentation encountered with Hypomelanosis of Ito may be the only clue to an underlying chromosomal mosaicism. Here, the skin pigmentation patterns can represent the distinct chromosomal admixture. Hemangiomata are also noteworthy, particularly those of the face and neck as they may signal the presence of other such vascular malformations affecting the airway.
The neurologic exam, while clearly helpful for diagnostic purposes may have added impact on an infant’s ultimate prognosis. Evaluation of tone, basic reflexes, involuntary movements, and seizure activity are critical to management of the patient [5].
An Overview of Genetic Testing
Chromosome Analysis
The field of genetics has experienced tremendous technological advances since the completion of the Human Genome Project in 2000. A brief review of techniques currently used in molecular diagnostics and their applications is discussed.
Methodology for identification and microscopic examination of the integrity of chromosomes became routinely available with the development of G-banding techniques in the 1960s. This staining methodology is still used today to identify aneuploidy involving whole chromosomes, or large (>5–7 Mb) rearrangements such as duplications and deletions within chromosomes. Standard G-banded karyotypes are typically performed on peripheral blood from the patient after stimulating the lymphocytes to grow and divide in culture. Usually, after 72 h in culture, a sufficient number of cells can be harvested to perform karyotyping. The process allows the cells to be in the metaphase stage of cell division when the chromosomes lengthen into distinct structures. Special Giemsa-based staining then allows the dark and light colored banding patterns to be discerned by further analysis by a cytogeneticist. This has been the standard technique used for detection and confirmation of common trisomies such as Trisomy 13, 18, or 21 or disorders of the sex chromosomes. Currently, it is still the only clinical diagnostic technique that can detect balanced translocations where chromosomal material from one chromosome is rearranged onto another. A balanced translocation does not result in a change in the dosage or copy number of a chromosomal region or of the genes residing in that region.
It is important to recognize that a normal karyotype (either from a prenatal CVS or amniocentesis specimen or from a postnatal blood sample) does not exclude the possibility of a chromosomal disorder. Due to the advent and rapid adoption of newer microarray technology described below, a large number of microdeletion/microduplication syndromes have been recognized, many of which are associated with congenital airway malformations. For example, the 22q11.2 microdeletion syndrome has been associated with cleft palate and laryngeal webs, which may lead to significant airway compromise during the neonatal period. Such submicroscopic gains and losses of chromosomal regions are considered copy number variations (CNVs). They may be detected within a chromosome (interstitial) or at the ends of the chromosome (subtelomere) and can result in change in dosage of a gene or genes located in a given region.
Fluorescence in situ hybridization or FISH is a molecular cytogenetic technique where a specific fluorescent-tagged DNA probe representing a single locus or part of a particular chromosomal region hybridizes to that region of the genome. It is useful in cases where the patient’s features are suggestive of a chromosomal disorder that is not typically visible under the microscope. Indeed, these submicroscopic genomic disorders have been, until recently, primarily identified by FISH and are associated with known clinical syndromes. The most frequently occurring microdeletion syndrome is the chromosome 22q11.2 deletion syndrome, associated with the clinical DiGeorge and Velocardiofacial (VCFS) syndromes. In addition, other chromosome-specific deletion syndromes such as those of chromosome 15q11.2 (Prader-Willi Syndrome and Angelman syndrome), chromosome 7q11.2 (Williams syndrome) have historically all been identified using a FISH probe for their respective regions. The disadvantage of this technique is that the clinician must know and specifically order the proper FISH test for a given chromosomal locus and many of the syndromes, even the well-recognized ones, can have atypical presentations or overlap with other diagnoses. In addition, in some cases, a patient may have a rearrangement that does not have the typical breakpoints associated with a given region and may not be detectable using the standard FISH probe. For these reasons, FISH has largely been replaced by chromosomal microarray analysis except for specific applications such as for testing family members of a proband.
FISH continues to be utilized for prenatal genetic testing. This modality can also be used to rapidly assess for trisomy or absence of whole chromosomes even without culturing and stimulating the cells toward metaphase. Such interphase FISH is routinely used as a rapid screen for aneuploidy of chromosomes 13, 18, 21, X and Y on chorionic villus sampling (CVS), amniocentesis, and in postnatal cases where rapid diagnosis may be desired for medical decision making, such as for an infant with features of Trisomy 13 or 18.
Over the last decade, single-locus testing by FISH has given way to chromosomal microarrays. This technique was originally based in comparative genome hybridization where patient DNA is labeled with one fluorescent probe while reference DNA is labeled in another and hybridized together. This allowed detection of regions across the genome, which had changes in copy number relative to the reference DNA and reflecting either gains (duplications) or losses (deletions) in the patient DNA. More recently, this approach has given way to the use of synthesized oligonucleotide probes or single nucleotide polymorphism (SNP)-based probes densely arranged on a microarray chip. Today, over two million such probes are routinely used on clinical diagnostic platforms for genome-wide postnatal testing. This allows the potential to report copy number changes in the 50–100 kb range. The processing is automated and copy number variation is compared against a large panel of standards. As might be expected with such high resolution analysis, changes that are of unknown significance are encountered and require further investigation and often examination of parental samples to help interpret the proband’s results.
Chromosome microarrays have provided a rapid, highly reproducible way to assess for submicroscopic chromosome rearrangements that might underlie an airway malformation, in an objective manner across the genome. It tests the regions associated with known syndromes and those not yet described, by identifying regions and groups of genes whose dosage has been changed due to the rearrangement. The widespread use of this technique for postnatal cases has led to the identification of several new genomic disorders. For these reasons, chromosome microarray has been recommended as the first-line study for cases of multiple congenital malformations and unexplained intellectual disability [6]. This recommendation has also been extended to prenatal cases of multiple congenital malformations [7].
The array techniques rely on the detection of copy number change relative to standards from arrays run on control individuals. Structural changes that do not result in copy number variation, such as a balanced translocation or inversion of part of a chromosome, will not be detected by this technique. For example, the presence of an extra copy of material from chromosome 21 will be detected on a microarray for a patient presenting with Down syndrome. However, whether this has occurred due to a trisomy (a free-standing extra chromosome 21) or from a translocation that might also occur in a balanced form and be carried by a healthy parent will not be differentiated by the microarray. Knowledge of the mechanism is necessary to provide an accurate recurrence risk for the parents and further testing and genetic counseling is indicated.
Single Gene Disorders. Many syndromes are due to the disruption of the normal function of a single gene or single gene disorder. An alteration of a single base pair in the sequence of the gene leads to a disruption in the gene’s functional product. In other cases, insertions or deletions of even a small number of nucleotides can disrupt the gene and subsequently, its protein product. These insertions or deletions may be as small as 2–3 base pairs, or may involve hundreds of base pairs. Neither a standard karyotype nor chromosomal microarray will detect these small sequence changes. These techniques may fail to detect deletions that encompass an entire gene, especially for small genes. Direct sequencing of the target gene will reveal single base pair mutations or small insertions and deletions. Larger deletions or duplications that involve either part or all of the gene are often even more difficult to detect, being too small to detect on a chromosomal microarray, but too large to detect with standard sequencing. These genetic lesions often require PCR-based deletion/duplication testing targeted to a specific gene locus. It is important to consider these potential mutational mechanisms and ensure they have been appropriately tested prior to eliminating a diagnosis from consideration.
Airway malformations can be part of a constellation of findings that result from mutation in a single gene. CHARGE syndrome is a good example, with mutations in the CHD7 gene associated with the clinical findings of CHARGE. Assessment of single gene disorders requires different techniques than those described above. Until recently, PCR-amplification and Sanger sequencing were the most commonly used techniques, requiring a gene-by-gene approach. These techniques are designed to examine the DNA at an individual nucleotide level assessing for alterations of one nucleotide for another (e.g., cytosine to adenine, C to A), deletion of one or more individual nucleotides or the insertion of a single or group of nucleotides into the normal sequence. The deletions or insertions have the potential to change the reading frame, resulting in a frameshift, which can affect production of the resulting mRNA and protein. A mutation that substitutes one nucleotide for another can result in formation of a premature termination codon and truncation of the putative protein. The mutation then is known as a nonsense mutation. An alteration in the DNA sequence that results in substitution of one amino acid for another in the translated protein, is known as a missense mutation, and can lead to production of a defective protein product. Gene sequencing can detect a variety of disease-causing mutations but requires that the clinician has selected the correct gene to test, which can be difficult in disorders with variable features.
Sequencing is not the testing method of choice for alterations where large segments of a gene are altered. For example, sequencing may miss cases where an entire exon of a gene is missing and not amplify properly to be sequenced. The exon deletion may be too small for accurate detection on a chromosome microarray. In these situations, specialized techniques involving quantitative PCR or MLPA may be used.
Standard PCR or polymerase chain reaction is commonly used to amplify desired segments of DNA; however, it is not typically quantitative or related to the amount of input DNA. Real-time PCR instead is a kinetic measurement, which assesses amplification in the early linear stages of the reaction and utilizes fluorescent labeling to quantitate products. The higher the copy number of a segment of DNA at the start, the more rapidly a significant increase in fluorescent product can be detected. MLPA or multiplex ligation-dependent probe amplification allows the relative quantitation of dozens of different PCR products of various lengths simultaneously. These two quantitative techniques allow for rapid assessment of copy number of a gene, part of a gene or a group of genes in a specific chromosome region. These diagnostic approaches require the selection of the appropriate target regions and unlike arrays, are not genome-wide. They are however, useful to test for intragenic deletions and duplications that might be below the threshold of reliable detection for microarray-based diagnostic testing and are routinely used in this capacity.
Sequencing
As mentioned, sequencing is the primary diagnostic modality to assess for mutations causing single gene disorders. Sequencing until recently has referred to Sanger-based sequencing of a single gene where typically, PCR is used to amplify the coding regions of a given gene, sequenced and then compared to the reference DNA. In addition, targeted sequencing, to assess a small region of a gene that is known to harbor a specific familial mutation for example, is also in routine use. Both of these techniques require the identification of a specific gene to test and in targeted sequencing, the familial mutation must be known. This has served as the classical method to identify single gene disorders such CHD7 mutations causing CHARGE syndrome for example, or one of the FGFR-based craniosynostosis syndromes. It requires examining the genes one at a time.
Over the last several years, methods for massively parallel sequencing or next-generation sequencing have allowed the availability of next-generation sequencing panels, where multiple genes in a disease pathway can be tested simultaneously. This approach works well for disorders where more than one gene in a developmental or biochemical pathway, when mutated, can cause disease and where clinical overlap in the phenotypes exists. In other words, it may be difficult to determine which of the group of genes to test based on clinical examination. Examples of well-known panels include those used for hypertrophic cardiomyopathy, non-syndromic hearing loss, Noonan syndrome (RAS/MAPK spectrum disorders), and craniosynostosis syndromes.
Over the last 3–4 years, next-generation sequencing has been applied to clinical exome sequencing. Whole exome sequencing (WES) targets the protein-coding regions or exons of the human genome. This includes approximately 20,000 genes and accounts for about 2 % of the total genome [8]. Despite the relatively small percentage of the genome sampled, this strategy is skewed toward the regions that directly result in producing functional protein and has rapidly moved from primarily research-based studies of related cohorts for a specific disease, e.g., Miller syndrome, to clinical diagnostic use and availability for unknown conditions [9]. It is available through commercial diagnostic laboratories.
For exome sequencing, the exons are targeted and captured from the patient’s DNA sample. Then the captured material is subjected to massively parallel sequencing. The patient’s sequence is then compared to reference DNA sequences, published control individuals and whenever possible, the patient’s family members. The typical scenario in a pediatric patient is to analyze a trio, or the patient and both parents to use as comparison. The analysis requires a series of sophisticated bioinformatics analyses often referred to as a pipeline. This reduces the thousands of variants that can be seen with exome analysis and filters the gene list down to the most likely ones. Clinical information and family history that may support a specific mode of inheritance are critical pieces of information for the interpretation of whole exome data. While whole exome sequencing provides an important new diagnostic test, it is not designed to detect larger chromosomal duplications and deletions that microarray is designed for and is limited to the protein-coding regions and splice junctions of the genes.
Whole genome sequencing instead,is designed using next-generation techniques to sequence both introns and exons providing more complete coverage than exome alone. This is, at this time, not routinely available for patient care though applications for cancer diagnosis seem imminent. As might be imagined, interpretation of these massive data sets will require significant bioinformatics and analysis capacity.
As noted above, it is possible to examine cell-free DNA in the maternal circulation that comes from a fetus. Currently analysis is primarily used to assess for aneuploidy of chromosomes 13, 18, 21, and the X and Y chromosome using a sequencing-based approach on the isolated cell-free DNA. This can take place as a peripheral blood draw as early as 10–11 weeks of gestation and is referred to as noninvasive prenatal testing (NIPT) or noninvasive fetal testing (NIFT). These screening tests are reported to have high sensitivity and are now being adapted to screen for common microdeletion syndromes. At this writing, while NIPT screening shows signs of rapid adoption, only invasive testing involving samples taken from CVS or amniocentesis is considered diagnostic and positive NIPT screens prompt one of these diagnostic studies.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


