The following tumors are classified based on tissues of origin, including choristomatous, epithelial, melanocytic, vascular, fibrous, xanthomatous, and lymphoid/leukemic origin.
Choristomatous Conjunctival Tumors
A variety of tumors can be present at birth or become clinically apparent shortly after birth. Most of the lesions are choristomas, consisting of displaced tissue elements normally not found in these areas. A simple choristoma is comprised of one tissue element such as epithelium, whereas a complex choristoma represents variable combinations of ectopic tissues like bone, cartilage, and lacrimal gland.
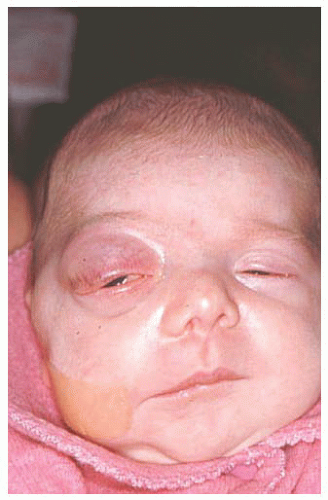
Dermoid Conjunctival dermoid is a congenital wellcircumscribed yellow-white solid mass that involves the bulbar or limbal conjunctiva (
25,
26). It characteristically occurs inferotemporally and often this tumor has fine white hairs (
Fig. 19.6). In rare cases, it can extend to the central cornea or be located in other quadrants on the bulbar surface. Most often dermoid straddles the limbus, but in rare cases it can be extensive and involves the full-thickness cornea, anterior chamber, and iris stroma. The more severe dermoids occur earlier in embryogenesis.
The conjunctival dermoid can occur as a solitary lesion or can be associated with Goldenhar’s syndrome. The patient should be evaluated for ipsilateral or bilateral preauricular skin appendages, hearing loss, eyelid coloboma, orbitoconjunctival dermolipoma, and cervical vertebral anomalies.
Histopathologically, the conjunctival dermoid is a simple choristomatous malformation that consists of dense fibrous tissue lined by conjunctival epithelium with deeper dermal elements including hair follicles and sebaceous glands. The management of an epibulbar dermoid includes observation if the lesion is small and visually asymptomatic. Anterior segment OCT can assist in judging depth of involvement. It is possible to excise the lesion for cosmetic reasons, but the remaining corneal scar can be cosmetically unacceptable. Larger or symptomatic dermoids can produce visual loss from astigmatism. These can be approached by lamellar keratosclerectomy with primary closure of overlying tissue if the defect is superficial, or closure using corneal graft if the defect is deep or of full thickness. The cosmetic appearance might improve, but the refractive and astigmatic error and visual acuity might not change. When the lesion involves the central cornea, a lamellar or penetrating keratoplasty is necessary and long-term amblyopia should be anticipated.
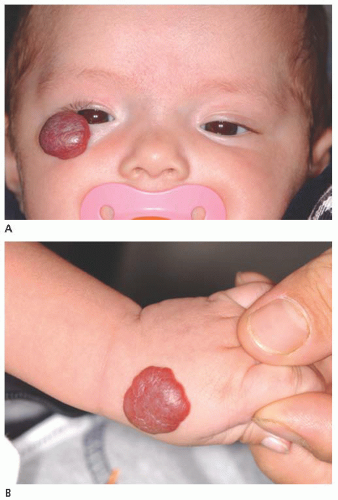
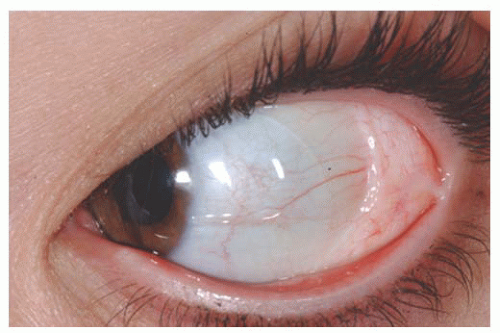
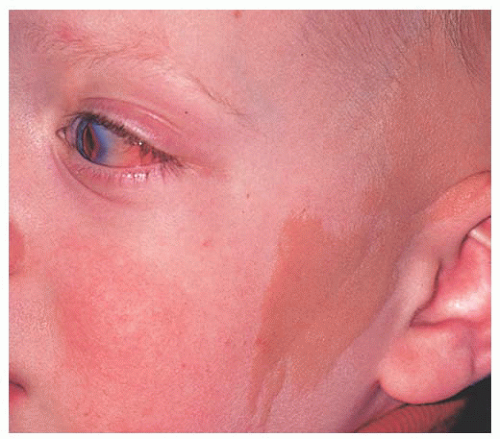
Dermolipoma Dermolipoma is believed to be congenital, but it classically remains asymptomatic for years and might not be detected until adulthood. This tumor tends to occur in the conjunctival fornix superotemporally and appears as a yellow, soft, fluctuant mass with fine white hairs on its surface (
Fig. 19.7). It can extend into the orbital fat and onto the bulbar conjunctiva, sometimes reaching the limbus.
Dermolipoma has features similar to orbital fat on CT and MRI scans. Histopathologically, it is lined by conjunctival epithelium on its surface and the subepithelial tissue has variable quantities of collagenous connective tissue and adipose tissue. Pilosebaceous units and lacrimal gland tissue might be
present. The majority of dermolipomas require no treatment, but larger ones or those that are cosmetically unappealing can be managed by excision of the entire orbitoconjunctival lesion through a conjunctival forniceal approach or by removing the anterior portion of the lesion in a manner similar to that used to remove prolapsed orbital fat. Amniotic membrane graft might be necessary to repair the defect.
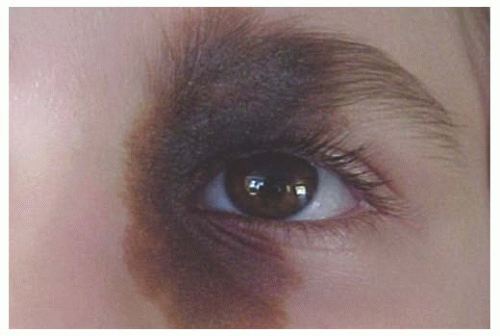
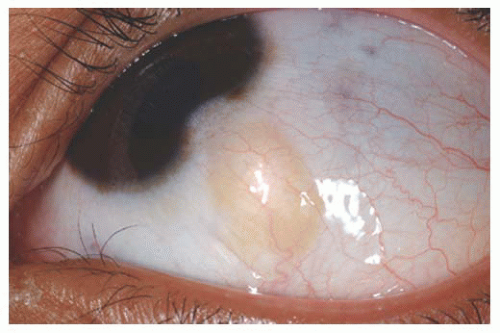
Epibulbar Osseous Choristoma Epibulbar osseous choristoma is a tumor comprised of mature bone, usually located in the bulbar conjunctiva superotemporally (
3,
27) (
Fig. 19.8). It is believed to be congenital and typically remains undetected until palpated by the older patient. On ultrasonography or CT scanning, the mass demonstrates calcium. This tumor is usually managed by observation. Occasionally, a foreign body sensation necessitates excision of the mass using a conjunctival forniceal incision followed by dissection of the tumor to bare sclera.
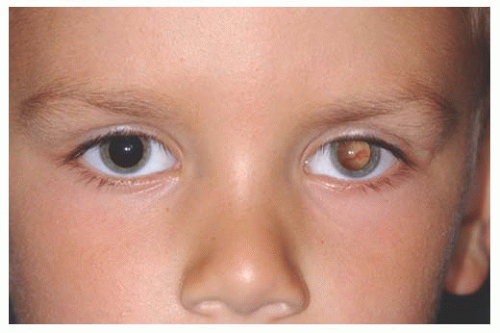
Lacrimal Gland Choristoma Lacrimal gland choristoma is a congenital lesion, discovered in young children as an asymptomatic pink stromal mass, typically in the superotemporal or temporal portion of the conjunctiva. It is speculated that this lesion represents small sequestrations of the embryonic evagination of the lacrimal gland from the conjunctiva. The lacrimal gland choristoma can masquerade as a focus of inflammation due to its pink color. Rarely, this mass can be cystic due to ongoing secretions if there is no connection to the conjunctival surface. Excisional biopsy is usually performed to confirm the diagnosis.
Complex Choristoma The conjunctival dermoid and epibulbar osseous choristoma are simple choristomas as they contain one tissue type such as skin or bone. A complex choristoma contains a greater variety of tissue derived from two germ layers such as lacrimal tissue and cartilage. It is variable in its clinical appearance and can cover much of the epibulbar surface, or it may form a circumferential growth pattern around the limbus.
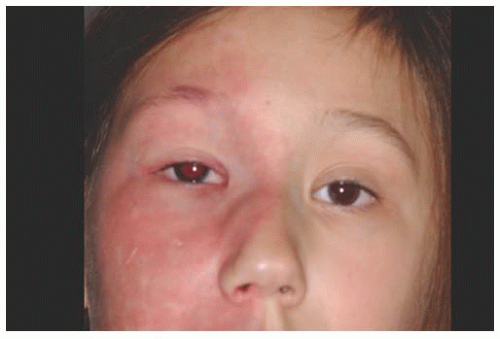
The complex choristoma has an association with the linear nevus sebaceous of Jadassohn (
26) (
Fig. 19.9). The nevus sebaceous of Jadassohn includes cutaneous features with sebaceous nevus in the facial region and neurologic features including seizures, mental retardation, arachnoidal cyst, and cerebral atrophy. The ophthalmic features of this syndrome include epibulbar complex choristoma and posterior scleral cartilage. The management of complex choristoma depends upon the extent of the lesion. Observation or wide local excision followed by mucous membrane graft reconstruction are options.
Epithelial Conjunctival Tumors
There are several benign and malignant tumors that can arise from the squamous epithelium of the conjunctiva.
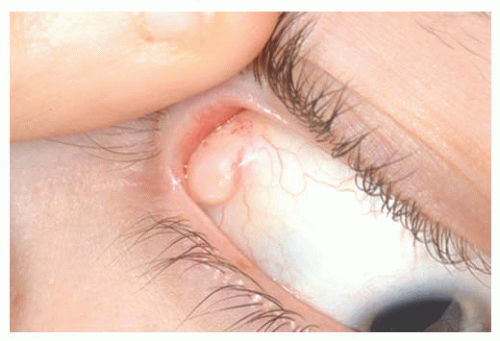
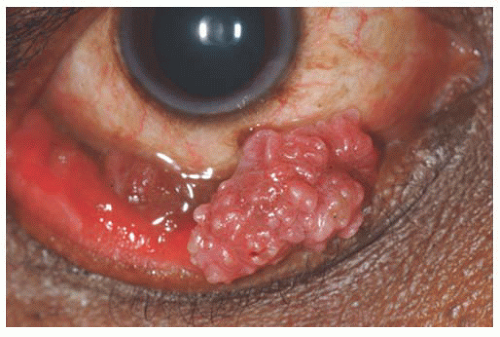
Papilloma Squamous papilloma is a benign tumor, documented to be associated with human papillomavirus infection of the conjunctiva (
28,
29). This tumor can occur in both children and adults. It is speculated that the virus is acquired through transfer from the mother’s vagina to the newborn’s conjunctiva as the child passes through the mother’s birth canal. Papillomas appear as a pink fibrovascular frond of tissue arranged in a sessile or pedunculated configuration (
Fig. 19.10). The numerous fine vascular channels ramify through the stroma beneath the epithelial surface of the lesion. In children, the lesion is usually small, multiple, and located in the inferior fornix. Histopathologically, the lesion shows numerous vascularized papillary fronds lined by acanthotic epithelium.
There are several treatment options for small sessile papillomas. Sometimes observation allows for slow spontaneous resolution of this tumor. Larger or more pedunculated
tumors can lead to foreign body sensation, chronic mucous production, hemorrhagic tears, incomplete eyelid closure, and poor cosmetic appearance, requiring therapeutic intervention. Complete removal of the mass without direction manipulation of the tumor (no touch technique) is advisable to avoid spreading of the virus (
30). Double freeze-thaw cryotherapy is applied to the remaining conjunctiva around the excised lesion in order to prevent tumor recurrence. In some instances, the pedunculated tumor is frozen alone and then excised while frozen. Topical or injection interferon and Mitomycin C have been employed for resistant or multiply recurrent conjunctival papillomas (
31,
32). For difficult recurrent lesions, oral cimetidine for several months following surgical resection can minimize recurrence by boosting the patient’s immune system and suppressing the virally stimulated mass (
33).
Hereditary Benign Intraepithelial Dyskeratosis Hereditary benign intraepithelial dyskeratosis (HBID) is a rare, benign condition seen in an inbred isolate of Caucasian, African American, and Native Americans (Haliwa Indians), initially identified in North Carolina. It is an autosomal dominant disorder characterized by bilateral elevated fleshy plaques on the nasal or temporal perilimbal conjunctiva and on the buccal mucosa. It can remain asymptomatic or can cause redness and foreign body sensation. It is characterized histopathologically by acanthosis, dyskeratosis on the epithelial surface and deep within the epithelium, and prominent chronic inflammatory cells. HBID does not usually require aggressive treatment. Smaller, lesssymptomatic lesions can be treated with ocular lubricants and topical corticosteroids. Larger symptomatic lesions can be managed by local resection with mucous membrane grafting if necessary.
Squamous Cell Carcinoma/Conjunctival Intraepithelial Neoplasia Squamous cell carcinoma and conjunctival intraepithelial neoplasia (CIN) are malignancies of the surface epithelial cells. Intraepithelial neoplasia displays anaplastic cells within the epithelium, whereas squamous cell carcinoma displays extension of anaplastic cells through the basement membrane into the conjunctival stroma. Clinically, invasive squamous cell carcinoma is usually larger and more elevated than CIN. Leukoplakia can be seen with either condition.
Patients who are medically immunosuppressed from organ transplantation, those with HIV, or those with underlying DNA repair abnormalities like xeroderma pigmentosum are at particular risk to develop conjunctival squamous cell carcinoma and malignant melanoma (
34). In these cases, the risk for life-threatening metastatic disease is greater.
The management of conjunctival squamous cell carcinoma varies with the extent of the lesion. Tumors in the limbal area require alcohol epitheliectomy for the corneal component and partial lamellar scleroconjunctivectomy with wide margins for the conjunctival component followed by freeze-thaw cryotherapy to the remaining adjacent bulbar conjunctiva. Extensive tumors or those tumors that are recurrent, especially with extensive corneal component, are treated with adjuvant topical Interferon, Mitomycin C, or 5-Fluorouracil (
31,
32,
34,
35).