Ocular Toxoplasmosis
Gary N. Holland
G. Richard O’Connor
Surveys have consistently shown ocular toxoplasmosis—caused by infection with the protozoan parasite Toxoplasma gondii—to be the most common form of posterior uveitis in many parts of the world, found in at least 30% to 50% of all cases.1,2,3 The rate varies, depending on the geographic area in which a given survey is made, age of patients examined, and other important factors such as dietary habits and immunologic status. Most ocular disease episodes that are encountered by ophthalmologists represent recurrent disease. The source of the original infection is not always known, and concepts regarding primary retinal infections are in evolution.
The lesions of ocular toxoplasmosis (Fig. 1) can be important causes of blindness, particularly if they involve the macula, the papillomacular bundle, or the optic nerve. Occasionally, a large peripheral lesion casts off so much inflammatory material into the overlying vitreous humor that vision is affected. In some cases, inflammation initiates organization of the vitreous body; on subsequent contraction of the fibrous bands within this inflammatory mass, tearing of the retina may occur and retinal detachment may ensue.
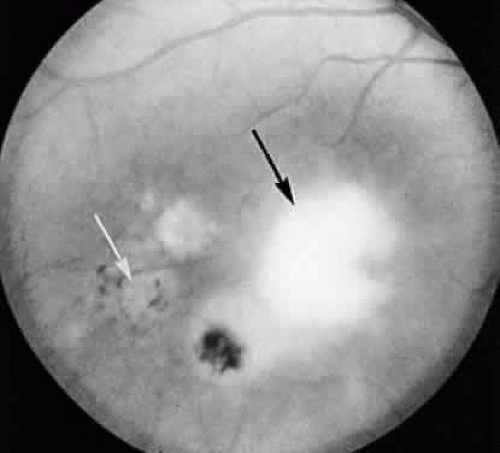 Fig. 1. The characteristic lesions of toxoplasmic retinochoroiditis in the fundus of an adult. Black arrow denotes active lesion with indistinct borders. White arrow denotes healed “satellite lesions.” (O’Connor GR: Ocular toxoplasmosis. In Locatcher-Khorazo D, Seegal BC (eds): Microbiology of the Eye. St Louis, CV Mosby, 1972.) |
If humans are to be spared the effects of ocular toxoplasmosis, the answers must be found to certain pressing questions. How do humans acquire the disease? Can truly effective methods of prevention be found: in particular, can effective vaccines be developed that can be given at an early age? What elements of the antigenic structure of T gondii are important in producing effective immunity? Once infection in the eye is established, what can be done to minimize effects? What is the relative importance of hypersensitivity to the organism or to its antigenic products in the damage that occurs in recurrent episodes? If recurrent attacks are the result of the release of encysted organisms into normal retinal tissue, how can the tissue cysts be eradicated? What are the most suitable antimicrobial agents for treatment of ocular toxoplasmosis: when and for how long should they be administered? These and a host of other questions remain for researchers to answer.
This chapter emphasizes information about the epidemiology of toxoplasmosis and about associated disease mechanisms that will help clinicians to evaluate patients appropriately for ocular involvement and to manage ocular lesions effectively. More detailed information about the clinical features of disease, diagnostic tests, and drug treatment is available in other references.4,5,6
PREVALENCE
Toxoplasmosis is virtually worldwide in distribution. Serologic surveys have shown a high prevalence of antibodies in tropical areas such as the South Pacific and a relatively low prevalence of antibodies in cold regions such as Iceland.7,8 At any given latitude, those living in mountainous areas above altitudes of 5000 feet seem to show lower degrees of infestation than racially and gendermatched counterparts living at sea level, but the reason for this difference is unclear.
The true prevalence of ocular toxoplasmosis in various communities is not known for several reasons. Ocular disease may be asymptomatic or minimally symptomatic, and scars may exist in individuals who had remote episodes of toxoplasmic retinochoroiditis that went unrecognized. Ocular toxoplasmosis may occasionally be confused with other forms of retinitis and uveitis or even with neoplastic disorders, such as large-cell lymphoma.
A large community survey in the state of Maryland identified retinochoroidal scars consistent with healed lesions of ocular toxoplasmosis in 0.6% of the population, and additional studies have found similar rates in other parts of the United States.9 In contrast, nearly 18% of the population in some areas of southern Brazil have such scars.10
If an expectant mother acquires systemic toxoplasmosis during the course of her pregnancy, she is at substantial risk (about 40%) for transmitting the disease to her unborn child.11 Based on numerous studies, the rate of congenital infections has been reported to be as low as 0.01% of live births in the United States to as high as 0.33% of live births in France.5 The child may appear to be healthy at birth but can develop signs of ocular or cerebral disease later. As many as 80% of individuals with congenital toxoplasmosis develop retinal lesions but these lesions may not develop for months or years after birth.12,13 Routine surveys of institutions for mentally defective children have uncovered the telltale signs of healed retinochoroiditis in the fundi of many of the institutionalized individuals, indicating that congenital toxoplasmosis—detected or not detected at birth—was the cause of mental retardation and eye lesions.14 Ocular lesions may be the only manifestation of congenital infection, however.
For many years, the rate of ocular involvement in patients with acquired T gondii infection was believed to be in the range of 2% to 3%.15,16 More recently, it has been shown that the rate may be higher. In 1995, contaminated water from a municipal reservoir was implicated in an epidemic of acquired toxoplasmosis in Victoria, British Columbia.17 After the epidemic, 100 cases of acquired toxoplasmosis were identified. Of the 100 patients, 19 had active retinitis, consistent with T gondii infection.
The frequency of recurrent attacks of inflammation is highly variable and cannot be foretold. Observed rates of recurrent disease are lower than the predicted prevalence of toxoplasmic lesions in a population.18 The median age at which recurrence develops has been reported to be early in the third decade, with 75% of reported cases occurring between 10 and 35 years of age.5,19
Ocular toxoplasmosis appears to be more common among immunosuppressed patients than in the general population. In patients with acquired immunodeficiency syndrome (AIDS), ocular involvement is less common than toxoplasmic encephalitis, however.
THE PARASITE
Toxoplasma gondii is an obligate intracellular protozoan parasite. Its exact place in the world of protozoan parasites was not determined until 1970, when Hutchison and colleagues20 in Scotland and Frenkel and coworkers21 in the United States discovered almost simultaneously the coccidian nature of this organism. Coccidian parasites undergo sexual reproduction in epithelial cells of the small intestine. The taxonomic classification of T gondii is as follows:
Phylum: Protozoa
Subphylum: Apicomplexa
Class: Sporozoasida
Subclass: Coccidiasina
Order: Eucoccidiorida
Suborder: Eimeriorina
Family: Sarcocystidae
Subfamily: Toxoplasmatinae
Genus: Toxoplasma
Species: gondii (only one species known).
The one-celled nature of the parasite justifies its designation as a protozoan. The organism, which measures about 7 μ in length by 2.5 μ in width, has a blunt posterior and a more pointed anterior end. Anteriorly, a “conoid” or primitive mouth is encountered. The conoid contains several internal organelles, including a polar ring from which 22 subpellicular tubules originate (Fig. 2). Rhoptries—specialized organelles that seem to have a secretory function—also terminate in this area, which is known as an apical complex. Hence, the subphylum is called “apicomplexa.” T gondii invades its host by means of sporozoites, eight of which are ultimately derived from each fertilized macrogametocyte, or ookinete. The crescent-shaped sporozoites penetrate the host cell membrane with their conoids, justifying the class designation “Sporozoasida.” Members of this class have both sexual and asexual reproduction. They move by gliding and flexing.
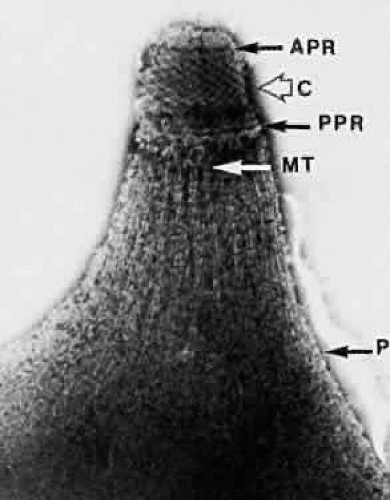 Fig. 2. Electron micrograph of the apical complex of a T gondii tachyzoite. Subpellicular structures have been revealed by negative staining with phosphotungstic acid (× 82,000). APR, anterior polar ring; C, conoid; PPR, posterior polar ring; MT, microtubule; P, remnant of pellicle. |
The discovery that sexual reproduction of T gondii takes place in the intestinal epithelium of cats immediately before shedding of oocysts in the feces led to its assignment to the subclass Coccidiasina. Members of this subclass are almost exclusively parasites of vertebrates and their gametocytes are intracellular.
T gondii, similar to certain other sporozoan parasites including Sarcocystis and Besnoitia species, forms cysts in certain tissues of the host, notably skeletal muscle or brain. These tissue cysts, containing hundreds or thousands of slowly multiplying parasites (bradyzoites), are the product of asexual reproduction. The tissue-cyst walls are derived partly from materials secreted by the parasite and partly from materials that come from the parasitized host cell. The ingestion of animal flesh infested with these tissue cysts facilitates the transfer of the parasite to a new host; the tissue cyst is relatively resistant to peptic digestion in the stomach but is readily susceptible to the action of trypsin in the small intestine. T gondii, resembling Sarcocystis species regarding the formation of tissue cysts derived from asexual reproduction, is considered to be a member of the family Sarcocystidae. All members of this family are monoxenous; that is, their definitive host is a single vertebrate species, such as the cat. All reproduce by a unique form of cell division termed endodyogeny.
Lastly, the subfamily Toxoplasmatinae, named after its prototype Toxoplasma species, has many distinct characteristics. Its sporocysts are contained within thin membranes in the oocyst. Both meronts (schizonts) and gamonts (sexual forms) are found within intestinal cells, and the gamonts mate there to form oocysts.
LIFE CYCLE
As described, T gondii exists in several different biologic forms. On its release from a sporulated oocyst, the sporozoite may multiply rapidly in intestinal epithelium for several generations before migrating through the intestinal wall and invading regional lymph nodes; this rapidly multiplying form is called a tachyzoite. Reproduction of the tachyzoite may occur by schizogony, the simultaneous cleavage of nuclear and cytoplasmic materials to yield several equal-sized merozoites that usually adhere to each other in the form of a “rosette.”
Some tachyzoites migrate within the protective milieu of leukocytes to extraintestinal sites, including skeletal muscle, heart, brain, and eye. Here, because of biochemical signals that are as yet imperfectly understood, they become encysted and slowly begin the process of bradyzoite (slow cell) formation. Conditions of cell nutrition, including the availability of the products of oxidative metabolism, may influence the shift to tissue-cyst formation. In contrast, Shimada and associates22 showed that the presence of both specific anti-T gondii antibody and complement in the immediate environment of cultured cells infected with T gondii stimulated the formation of tissue cysts. The rate of reproduction of cells in the mature retina and brain is slow. It may be that certain metabolic factors in the retina and brain favor the formation of tissue cysts in situ after the initial establishment of infection in these tissues. The wall of the tissue cyst is composed of complex proteins and polysaccharides arranged to permit the passage of water, gases, electrolytes, and amino acids across the tissue-cyst-wall barrier but substances of higher molecular weight, such as proteins, are probably excluded.
Tissue-cyst formation at these sites is a potential disadvantage for the host because tissue cysts are reservoirs of slowly multiplying organisms that on rupture of the tissue cyst wall can cause a recurrence of inflammation. Organisms within tissue cysts are also harder to kill with drugs than are tachyzoites. Tissue cysts are probably the means by which most T gondii infections are passed from animal to animal. On eating the carcasses of chronically infected animals, birds and rodents become intermediate hosts for the parasite. When they in turn are eaten by cats, the life-cycle may be completed. If a cat has never been infected, the life-cycle may be initiated in the cat’s intestine.
The factors that influence merozoites (products of asexual reproduction) to become gametocytes are unknown but this conversion can occur only in the intestinal epithelium of the cat (including virtually all members of the cat family—Felidae). The cat is thus the definitive host for T gondii, and all other animal species that become infected with this parasite can be considered incidental or intermediate hosts. The macrogametocytes (female forms) and microgametocytes (male forms) each have their own highly distinctive morphologic characteristics, as demonstrated by Hutchison and associates.20 Microgametocytes generally produce about 12 microgametes, each of which penetrates a mature macrogamete. These forms unite within the superficial epithelium of the cat’s intestinal mucosa. This union produces an ookinete, which is subsequently shed in the cat’s feces as an unsporulated oocyst. Such oocysts require from 1 to 5 days for sporulation in an environment such as moist soil. Once sporulation has taken place, the oocysts are fully infective to humans in addition to numerous other species including herbivores (e.g., sheep, cattle). When previously uninfected cats are fed sporulated oocysts, the interval between the time of feeding and first appearance of oocysts in the cats’ feces is about 20 to 24 days.23 This “prepatent” period differs according to the form of T gondii that is fed to the cat. Oocysts can first be detected in the feces of cats 3 to 5 days after the feeding of T gondii tissue cysts and 5 to 10 days after the feeding of tachyzoites. Oocysts continue to be shed in the feces of infected cats for 7 to 20 days, with the average length of this “patent” period being about 12 days. Thus, a cat that is shedding oocysts in its feces may be a source of infection to humans and to other animals in its immediate environment but the total period of the cat’s infectivity is only about 2 weeks. The families of patients with ocular toxoplasmosis, having heard that the cat is the definitive host of T gondii, often ask whether they should get rid of their household pet. The folly of such a proposal is realized when it is understood that the cat in question probably ceased being infective months or even years before the patient developed eye disease.
Tissue cysts contained in the skeletal muscle or brains of intermediate hosts resist peptic digestion after ingestion by predators but are eventually broken down by tryptic digestion in the small intestine of the cat; the remainder of the life-cycle proceeds as described. Fayer24 has stated that more than 350 species of warm-blooded intermediate hosts of T gondii have been identified; in addition, a few cold-blooded species such as reptiles can act as intermediate hosts. The cat, on eating the flesh of any of these animals may become infected but it should be emphasized that cats kept within an ordinary urban environment and fed only processed foods have a smaller chance of becoming infected.
TRANSMISSION AND PATHOGENESIS OF HUMAN INFECTIONS
In areas such as Micronesia, where T gondii antibodies are widely prevalent even among young children, women of child-bearing age are presumably already immune as a result of asymptomatic childhood infections. Their first contact with the disease is never during pregnancy; thus, they never transmit T gondii to infants in utero. Interestingly, ocular toxoplasmosis is rarely seen in such populations.
Asymptomatic infection of humans is common throughout most of the temperate and tropical areas of the world. Correlations have been made between high rates of infection and the tendencies of certain segments of the world’s population to eat raw or undercooked meats, which is believed to be the most common mode of transmission.25 Most animals that are commonly used for domestic meat production have been shown to be infested with T gondii. The level of infestation among hogs and sheep is particularly high but cattle are also known to be infected. Masur and coworkers26 have described a minor epidemic of systemic toxoplasmosis within a family, all of whom had eaten rare lamb at one particular time.
Infection in humans can also be established by the ingestion or possibly the inhalation of sporulated oocysts. In the Victoria epidemic, water from a municipal reservoir appeared to be the vector. Another smaller epidemic of systemic toxoplasmosis affected 37 patrons of a particular riding stable near Atlanta, Georgia.16,27 These patients had not consumed food or water from a common source but T gondii oocysts were found in the dust that covered the floor of a particular part of the stable that had been frequented by feral cats.
Transmucosal infection has been postulated to occur, particularly in laboratory infections. The subject has been extensively reviewed by O’Connor.28 Appel and associates29 described submandibular adenopathy in a young girl who was believed to have been infected through the oral mucosa, and Räisänen30 showed the ease with which T gondii tachyzoites could penetrate the nasal and oral mucosa of mice, establishing fatal toxoplasmosis in some of these experimental animals. Transmission is also possible through colostrum or milk, as was first observed in suckling mice by Eichenwald in 1948.31 Rieman and coworkers32 subsequently demonstrated systemic toxoplasmosis in an infant who had been fed unpasteurized goat’s milk, and Sacks and associates33 described a small epidemic of systemic toxoplasmosis in a family that had partaken of raw milk from a chronically infected goat. In all of these cases, it seems that the T gondii tachyzoite was the culprit but the exact site of entry of the organism has not been determined. The highly acid environment of the stomach would be expected to kill tachyzoites after brief contact, unless the milk in the immediate environment of the parasites had sufficient buffering capacity to prevent such damage. Despite the relatively rapid transit of the infected milk over the oral and pharyngeal mucosa, transmucosal infection cannot be ruled out because Nichols and O’Connor34 have shown that T gondii tachyzoites can penetrate cells within 15 seconds of contact.
Although tachyzoites have been identified in saliva, transmission by kissing does not appear to be of importance.35 Price36 compared the prevalence of anti-T gondii antibodies in 43 married couples. In 23 of these couples, both partners were either seropositive or seronegative; in 20 couples, one member remained persistently negative, whereas the other was persistently positive. Some change from seronegativity to seropositivity would have been expected if transmission had occurred by kissing or other sexual contact. Frenkel and Wallace37 used this observation to argue against the hypothesis that trophozoites are transmitted across mucosal surfaces.
From the evidence, it appears that T gondii tissue cysts, tachyzoites, and oocysts are all capable of initiating systemic disease in humans. It is likely that the ingestion of tissue cysts contained in undercooked meat is the most frequent cause of disease and that tachyzoites and oocysts have only a relatively minor role. In the context of ocular disease, the transmission of T gondii tachyzoites across the placental barrier is an important mode of infection but may not be the predominant source of retinal lesions, as traditionally assumed.
CONGENITAL TOXOPLASMOSIS
Antibody-negative women of child-bearing age are at risk for acquiring T gondii infection and for transmitting the parasite to their fetuses during dissemination of the organism. The rate of transmission to the fetus actually is higher during the later months of pregnancy than it is during the first trimester, possibly because of the greater vascularity of the placenta in the later months. The amount of damage to the fetus is substantially greater when infection occurs in the first trimester, however, presumably because of the immaturity of the fetal immunologic defense system and the relatively greater vulnerability of fetal organs such as the brain and the eye during the first trimester. Infants born of women who acquire toxoplasmosis during pregnancy may appear to be perfectly normal at birth yet may develop signs of retinochoroiditis in later childhood, adolescence, or early adulthood. Generally, a woman who has given birth to one child with congenital toxoplasmosis will never give birth to another congenitally infected child.
Women are at little or no risk for transmitting the infection to their fetuses if they have been infected with T gondii previously because of the protective effects of antibodies. It appears, however, that maternal infections acquired within the last several weeks before conception can be transmitted to the fetus, even in immunocompetent women.38
In one study, macular lesions were present in 58% of children with ocular disease attributable to congenital toxoplasmosis.39 Macular involvement appears to be more frequent than would be predicted by the small area of the anatomic macula relative to the rest of the fundus.
ACQUIRED TOXOPLASMOSIS
Most individuals who acquire T gondii infection do not develop clinically apparent ocular infections. In the Atlanta epidemic, only one of the patrons of the riding stable (an 11-year-old girl) developed unilateral focal retinochoroiditis characteristic of acquired toxoplasmosis after 4 years of follow-up.16 Her acute, febrile, lymphadenopathic illness originally was no different from that of any of the other 36 patients. The more recent Victoria epidemic shows that the rate of ocular infection can be higher, however. Serologic studies also indicate that toxoplasmic retinochoroiditis may be associated with acquired infections more frequently than heretofore believed.40 Active toxoplasmic retinochoroiditis can develop months or perhaps even longer after the systemic illness associated with acquired infections.26
There has been much debate about whether characteristics of ocular lesions (e.g., bilaterality or location within the fundus) are different between acquired and congenital ocular infections.40,41 It appears, though, that generalizations are difficult to make and that both types of disease can be identical in appearance.
RECURRENT DISEASE
The timing of the primary infection in cases of recurrent ocular toxoplasmosis has been a subject of some controversy. In the past, there had been general agreement that nearly all recurrent toxoplasmic retinochoroiditis lesions represented a late relapse of congenital disease when tissue cysts were deposited in the retina during widespread dissemination of the parasite throughout the body. Holland and associates5 have summarized several lines of evidence used in the past to argue that retinochoroidal scars are almost always congenital in origin. For example, when T gondii infection is widespread in a population, first exposure usually occurs in childhood; therefore, infection during pregnancy with congenital transmission is uncommon. It has been observed that the rate of ocular toxoplasmosis is low in some areas of the world, such as Micronesia, where seropositivity rates are high.42 Perkins15 argued that if ocular lesions were attributable to acquired infection, the rate of recurrent disease should increase with age, as do rates of seropositivity. Recurrent ocular toxoplasmosis has been more common in the second and third decades rather than later in life, in some studies.19
More recent evidence, however, suggests that many of the scars from which recurrent inflammatory lesions arise were actually the result of infections acquired after birth.5 As stated, the rate of ocular involvement may be higher in cases of acquired infection than heretofore suspected. Ocular toxoplasmosis may be the only manifestation of acquired infection, and it may occur months after the infection is acquired.
Experience in southern Brazil differs from that in Micronesia.10,43 Congenital toxoplasmosis is uncommon, seropositivity rates are high, and the prevalence of ocular toxoplasmosis increases with age. Numerous nontwin siblings are found to have toxoplasmic retinochoroidal scars. These findings suggest that recurrent ocular toxoplasmosis may be related to acquired rather than congenital infections. The primary episodes in which scars were established might have been small and asymptomatic or might have occurred early in life, when children would not verbalize their visual symptoms.
In the Victoria epidemic, it has been estimated that the actual number of individuals infected was between 2894 and 7718—far more than the 100 who were identified.17 If the rate of ocular infection among all infected individuals was 19%, as in the group with known infection, then the epidemic may have caused hundreds of unrecognized ocular lesions. That numerous unrecognized ocular infections may have come from an unsuspected source (municipal drinking water) raises the possibility that primary lesions in many patients with recurrent inflammation may have been from a remote acquired infection rather than from congenital infection, as traditionally believed. Traditional and evolving concepts are both based on circumstantial evidence, and many of the observations and interpretations remain in conflict. It may be that a variety of host and parasitic factors determine the retinotropism of acquired infections, which would explain different observations in different populations. Additional study is required to resolve these questions.
Mechanisms by which lesion reactivation occurs are discussed in a subsequent section.
EXPERIMENTAL MODELS OF OCULAR TOXOPLASMOSIS
Many questions concerning the pathogenesis and therapy of ocular toxoplasmosis remain unanswered, which has led to efforts by various investigators over the past five decades to develop a satisfactory animal model of this disease. An ideal animal model of ocular toxoplasmosis should enable the investigator to produce lesions that closely resemble those of humans in morphology, clinical course, and histopathologic manifestation. The lesions should be readily reproducible in an eye that is anatomically and physiologically similar to a human eye. The immunologic defense system of the animal host should also be similar to the human system, and it should be possible to manipulate the immune response experimentally. The lesions should not be violently destructive, nor should they produce retinal detachment or severe lens opacity because these changes would preclude serial examination of the lesions. Also, experimentally induced infections should not cause such severe systemic reactions (e.g., fever, toxemia, encephalitis) as to induce premature death. To date, no model fulfills all of these criteria. Nevertheless, a better understanding of ocular toxoplasmosis has emerged from animal models that are available.
As early as 1951, Hogan44 recognized the need for a reliable animal model of ocular toxoplasmosis. On the assumption that T gondii probably gained access to the eye through the bloodstream, he injected living RH-strain T gondii parasites into the carotid artery of rabbits and produced focal chorioretinal lesions. The lesions could be seen by ophthalmoscopy within 3 or 4 days after inoculation but the natural course of the lesions could not be followed-up because the animals died of toxoplasmic encephalitis a few days later. These early experiments, however, provided specimens for the histopathologic analysis of blood-borne ocular infections with T gondii. The model was not optimally useful, however, because the retinal vasculature of the rabbit eye does not extend far beyond the edges of the optic disc. The lesions that Hogan produced were primarily choroidal rather than retinal, as in humans, thereby limiting the usefulness of this model.
In 1968, Nozik and O’Connor45 published another description of experimentally induced toxoplasmic retinochoroiditis in the rabbit. Their technique required the injection of a small inoculum of Beverley-strain T gondii organisms into the suprachoroidal space under direct ophthalmoscopic observation. Focal retinochoroiditis appeared near the posterior pole of the fundus 4 to 6 days after inoculation. Moderate clouding of the vitreous humor was seen in association with the retinal lesions but the latter remained visible at all times. The Beverley strain of T gondii, being less virulent than the RH strain, did not kill the animals. Activity in the experimentally induced fundus lesions lasted 4 to 6 weeks, at the end of which time pigmented atrophic scars developed. Histologic studies of the healing lesions showed tissue cysts of T gondii in the superficial retinal layers, in addition to numerous lymphocytes and plasma cells. This model was used extensively to test various theories concerning the recurrence of toxoplasmic retinochoroiditis.46,47 It was also used to test the efficacy of various therapeutic regimens in shortening the course of disease or rendering the retina free of viable parasites.48,49 Several workers realized that even this model had considerable shortcomings, however. For example, the rabbit eye did not show signs of retinal vasculitis or papillitis, as commonly seen in human beings. Furthermore, the rabbit’s immunologic defense system differed considerably from its human counterpart—being, among other things, a great deal more sensitive to the effects of corticosteroids.
A series of studies by O’Connor and colleagues47,50,51,52,53 at the Francis I. Proctor Foundation, using a nonhuman primate model of toxoplasmic retinochoroiditis, have provided additional insights into disease mechanisms. Culbertson and associates50 produced retinochoroiditis in the rhesus monkey by the intraretinal inoculation of RH-strain T gondii organisms. The resulting lesions looked much like human lesions, and retinal vasculitis was a common complication. In subsequent experiments, this model was used to investigate the relative roles of proliferating organisms and hypersensitivity reactions in the tissue destruction that occurs with recurrent ocular toxoplasmosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


