Ocular Pharmacology of Antibacterial Agents
John E. Sutphin
Jeffrey M. Wells
The empiric selection of an antibiotic to treat an ocular infection is based on several factors including the likely organisms, their historic susceptibilities, the host, and the properties of the drug. In all serious infections, attempts to identify the organism through culturing, polymerase chain reaction, or other methods should be completed before initiating treatment. Relevant features of the host include the immune status, site of the infection, history of drug allergies, drug interactions, special concerns such as age or pregnancy, and the host’s ability to absorb and metabolize the drug. Features of the drug are its pharmacodynamics or interaction between the drug and the infecting organism, its pharmacokinetics or interaction between the host and the drug, its immunomodulatory and adverse effects, treatment costs, and concerns for antibiotic resistance.1
Immunomodulatory actions of antibiotics include direct effects on phagocytosis via opsonization or oxidative intermediates, on chemotaxis, on lymphocyte proliferation, on cytokine production, and on delayed hypersensitivity and natural killer cell activity. Antibiotics shown to be upregulating include the imipenems, cefodizime, and clindamycin. Downregulation is demonstrated by the tetracyclines, erythromycin, roxithromycin, cefodizime, rifampicin, gentamicin, teicoplanin, and ampicillin.2 The ideal antiinfective agent would combine an enhancement of the natural bacterial killing mechanism and downregulation of an exaggerated inflammatory response.3
Pharmacoeconomics compares the costs and consequences of antibiotic therapy in view of efficacy, safety, and health system expenditures.4 Cost factors are continually changing as drug patents expire and improved agents become available. Overall costs are related as much to the method of delivery as to the individual agents selected. The Endophthalmitis Vitrectomy Study not only showed the lack of effectiveness of intravenous therapy but also the characteristics of the infection indicating the need for more expensive vitrectomy in conjunction with intravitreal antibiotics.5
Bacterial resistance is due to a variety of mechanisms that aim at disrupting the antimicrobial mechanism of action (Fig. 1) Increasing resistance of ocular pathogens represents an important therapeutic challenge. Antibiotic resistance is caused, paradoxically, by both overuse and underuse. Especially in developing countries, resistance often develops when people stop taking their medication as soon as they feel better.6 In the United States, resistance more often stems from overuse. The Centers for Disease Control and Prevention estimates that as many as half of the antibiotics prescribed by office-based physicians each year to treat colds, coughs, and other viral infections are unnecessary.6,7 The result of the overuse of antibiotics for systemic use, ophthalmic use, and in poultry and livestock feed, is an increasing number of bacterial strains that are stronger than the antibiotics being used against them.
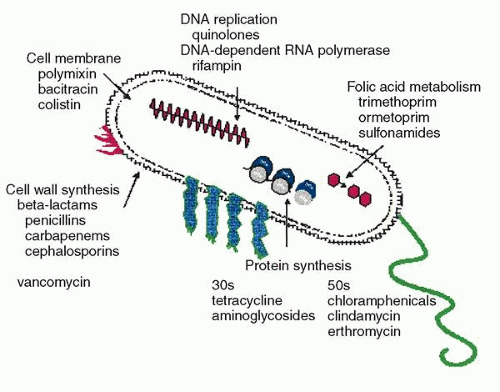 Fig. 1. Mechanisms of antibiotic resistance. (Modified with permission from Boothe DM: Small Animal Clinical Pharmacology and Therapeutics, p 141. Philadelphia, WB Saunders, 2001) |
PHARMACODYNAMICS
Pharmacodynamics is the quantitative relationship between observed plasma and/or tissue concentration of an active drug form and the pharmacologic effect. As opposed to pharmacokinetics, which studies how the body handles a drug, pharmacodynamics is the study of the effects of a drug on the microorganism and the pharmacologic effects of a drug in the body8
The minimal inhibitory concentration (MIC) is the lowest concentration of an antimicrobial agent that prevents visible growth of bacteria after a period of incubation in vitro.9 The MIC90 is the concentration at which 90% of the isolates by genus and species are inhibited (not killed). The MIC90 is ideally based on at least 100 organisms.10 For antimicrobial therapy to be effective, it is believed that a drug should, at a minimum, reach a concentration that at least exceeds the MIC90 of the target organism.
The breakpoint MIC (MICBP) of a drug is a yardstick against which the MIC for an infecting organism can be compared. The MICBP is that MIC separating susceptibility from resistance and is typically the concentration of a drug that is achieved reasonably in the serum or tissue. An organism is considered susceptible (S) if its MIC is one sixteenth to one fourth the peak drug concentration that will be achieved using the recommended dose of a drug, but this should be correlated with clinical response to the selected antimicrobial.10,11 If the MIC of the organism equals or surpasses the MICBP, the organism is considered resistant (R).11 The breakpoints in defining bacterial resistance are based on what can be achieved with systemic therapy, and therefore they may not always reflect ocular levels.
The minimal bactericidal concentration (MBC) is the lowest concentration of antimicrobial that totally suppresses growth on antibiotic-free media or that results in a 99.9% (3 log) reduction or greater decline in colony counts after overnight incubation. The MIC may be the same as the MBC, but the MBC can be several multiples of the MIC, in which case the organism is said to be resistant.9
Bactericidal drugs are defined as drugs whose MIC is very close to the minimal bactericidal concentration. However, the bactericidal effects of a drug depend on achieving sufficient concentration of the drug at the antimicrobial target tissue.12 It is important to note that bactericidal drugs can be rendered bacteriostatic should insufficient concentrations be reached. A drugs whose MBC is more than 16-fold greater than the MIC is bacteriostatic.
The inhibitory quotient (IQ) is the ratio of the antibiotic concentration in the target tissue to the MIC90 of a particular organism.13 Generally, an IQ greater than 4 is preferred, because otherwise the antibiotic may inhibit the growth of the organism but not kill it. The concept of the “inhibitory quotient” links pharmacodynamics to pharmacokinetics but is susceptible to manipulation. For instance, the numerator is typically a mean tissue concentration, which does not reflect the high patient variability of 50% or more seen in drug penetration studies. The denominator can also be manipulated if strains are pre-selected or if several resistant strains are included. For this reason, development of an IQ expressed as a range of 25% to 75% might be a more accurate value than one number.
The relationship between bacterial killing and microbiological activity is described by the area under the curve (AUC) divided by the MIC of the organism (Fig. 2). This is known as the AUC/MIC ratio or area under the inhibition curve (AUIC). The AUC/MBC ratio is also used. Aminoglycosides and fluoroquinolones exhibit concentration-dependent killing; that is, bacteria succumb more rapidly when exposed to concentrations greatly in excess of MIC values than to concentrations just above their MIC. For β-lactams and glycopeptides (cellwall–active agents) this is not the case, and further increases to more than 4 to 8 times the MIC do not increase the rate of bactericidal kill.14 Not surprisingly, some studies note a plateau of fluoroquinolone induced killing rate at very high multiple of AUC/MIC.15
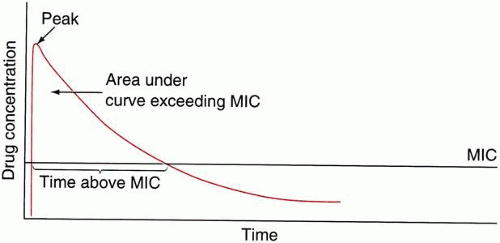 Fig. 2. Pharmacokinetic parameters related to the MIC (minimum inhibitory concentration). As one increases the AUC (area under the curve), the time above the MIC and the peak: MIC and AUC: MIC ratios all increase as well. (Reprinted by permission from Appleton & Lange, Stamford, CT: Principles of Therapeutic Drug Monitoring, third edition, by William E. Evans, Jerome J. Schentag, and William J. Jusko, published by Applied Therapeutics, Inc., Vancouver, Washington, 1992, Figure 97.11, p 1950.) |
Microbial killing of aminoglycosides and fluoroquinolones is most likely linked to high AUC/MIC ratios, but high peak concentrations may be important to suppress resistant mutants. If the peak concentration (Cmax) to MIC ratio is not high enough, a mutant subpopulation may emerge (16). Thus, using a dose that is too low or too infrequent is particularly detrimental with fluoroquinolones and aminoglycosides.
Time-kill curves11 are derived from in vitro susceptibility tests that determine both the overall killing capability of the antibiotic and the time required to kill. The end point is the MBC, which means that 99.9% of bacteria are killed, compared to the MIC90, in which 90% are inhibited (Fig. 3). The potential advantage of time-kill curves is that they add a time component to the measurement of bacterial killing. However, all too often, the full potential of this approach is not realized because of inappropriate experimental design and/or suboptimal quantitation of bacterial killing and regrowth curves, or the antimicrobial effect itself.
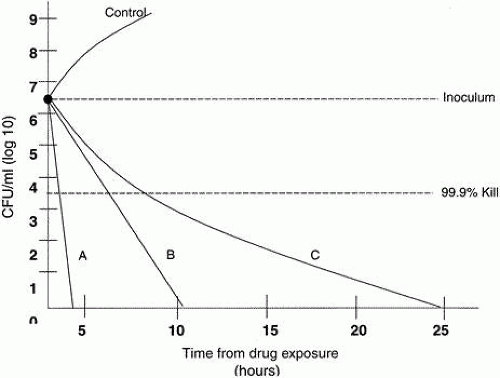 Fig. 3. Time-Kill Curve. Broth cultures inoculated with 5 × 106 CFU/mL are incubated and then exposed to different antibiotics or to a control solution. The graph above illustrates different time-kill curves for antibiotics A, B, and C. These antibioticsC could also represent different concentrations of the same antibiotic. The end point is the time to a bactericidal effect, which is considered a 3-log reduction or 99.9% kill. |
Specifically, most of these studies have simulated human pharmacokinetics over a narrow dose range. This may not be appropriate for comparing different drugs, especially if the antimicrobial concentrations are close to the minimum or maximum values. Many time-kill curve studies are flawed because they compare different antibiotics at the same concentration, thereby ignoring pharmacokinetic factors such as the different penetration levels of drugs. Time-kill curves can vary greatly for a given antibiotic depending upon the concentration studied. The one-dose nature of such studies does not provide evaluation of the equiefficient dose of a new drug (the dose of the new drug that produces the same effect observed with a reference drug at its usual dose). Finally, most studies do not include a sufficient duration of observation to cover the entire regrowth phase in time-kill curve studies.17
Persistence of antimicrobial effects after brief exposure to, or the lack of detectable concentrations of, an antimicrobial is termed the post-antibiotic effect (PAE).18 For some drugs (e.g., fluorinated quinolones and aminoglycosides), the duration of the PAE, and thus antibiotic efficacy, is concentration dependent and is maximized by a large peak drug concentration (Cmax) to MIC ratio.10 The PAE may also give a patient’s host defenses more time to recover, perhaps contributing to the absence of endophthalmitis in eyes with contaminated aqueous humor.
Very limited clinical data are available that could answer the question which of these pharmacodynamic variables are most important in the efficacy of ocular antibiotics under clinical conditions.16 Historically, the manner in which susceptibility is determined in vitro has been based on achievable serum levels of the drug that do not necessarily relate to ocular levels. The usual in vitro estimates of antimicrobial activity (MIC, MBC, and time-kill studies) are often determined at constant drug concentrations (static conditions) that do not consider pharmacokinetic parameters.17 Due to these limitations, in vitro data may not correlate with clinical outcomes.
The AUC/MIC ratio holds promise as a benchmark for comparing ophthalmic antimicrobial agents. In normal clinical practice, it is not possible to obtain a full concentration profile of a drug in the eye because of limited opportunity and the risks involved in the sampling of ocular fluids and tissues. A population pharmacokinetic approach (taking one sample per patient but at different times post-dose so that a curve can be plotted) may allow different agents to be compared and help us to better understand interpatient variability.19
Some clinicians are currently using AUC/MIC ratios as a measure for comparing different systemic antimicrobial agents. Since the interpretation of a value without correlation to outcome may be misleading, most clinicians rely primarily on evidence-based medical regimens obtained from clinical trials.20
PHARMACOKINETICS
Pharmacokinetics describes the quantitative relationship between the administered dose or dosing regimen and the observed plasma and/or tissue concentration of a drug. Pharmacokinetics can also be considered the study of what the body does to the drug.1
The eye is one of the few structures that can be considered pharmacokinetically separate from the body. This separation is possible because of two main factors: (1) absorption occurs directly into the eye before reaching the systemic circulation, and (2) elimination occurs directly into the body, which can be considered a large reservoir that can dilute drug concentration below a therapeutic threshold.21
There are five barriers to the entry of drugs into the eye (Fig. 4):
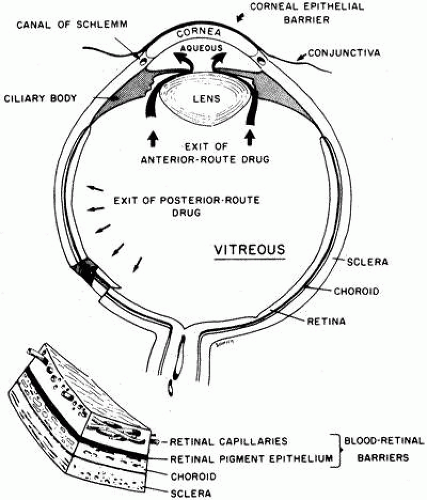 Fig. 4. Schematic diagram of the major pharmacokinetic features of the eye. There are three barriers to ocular penetration: the corneal epithelium, the blood-aqueous barrier (in the ciliary body), and the blood-retinal barriers. The outer blood-retinal barrier is in the retinal pigment epithelium; the inner one lies in the tight junctions of the retinal capillaries. Each contains an active transport pump for organic anions. Anterior-route drugs (aminoglycosides) leave the vitreous by way of the aqueous humor and canal of Schlemm. Posterior-route drugs (penicillins, cephalosporins) leave by active transport across the retina. (Barza M.: Pharmacokinetics of antibiotics. In Sabath LD (ed): Action of Antibiotics in Patients, p 29. Bern: Has Huber, 1982.) |
Corneal epithelium—Restricts the entry of water-soluble drugs into the cornea and aqueous humor. The barrier is breached by an epithelial defect or, if the epithelium is intact, is bypassed by subconjunctival injection.
Conjunctiva/sclera—Compared to the cornea, the conjunctiva and the anterior sclera have a much larger area and are more permeable to water-soluble drugs. However, drug loss by uptake into the conjunctiva is as important as preocular clearance in reducing the fraction of drug available for corneal absorption.
Aqueous-vitreous barrier—Bulk flow of aqueous humor from the eye and the presence of an intact lens and zonules retard the diffusion of drugs from the anterior chamber into the vitreous humor.
Blood-aqueous barrier—tight junctions of the iridal vasculature endothelial cells limit entry into the aqueous from the blood. The epithelium of the iris and ciliary body pump anionic drugs from the aqueous into the blood stream.
Blood-retinal barrier—tight junctions of the retinal vasculature limit the entry of drugs into the eye from the systemic circulation (internal). The pigment epithelial barrier also limits flow into the eye (external). There is an outward pumping of anions across the retina by the retinal pigment epithelium and the endothelial cells of the retinal vessels.22
In terms of drug delivery, the eye can be considered to have four target sites: (1) the pre-ocular structures of the front of the eye, such as the conjunctiva and eyelids; (2) the cornea; (3) the anterior and posterior chamber and associated tissues; and (4) the vitreous cavity. Topical administration to the front of the eye can be used to deliver pharmacologic agents to the preocular, corneal, and anterior/posterior sites, but this method is presently rarely used to deliver drugs to the vitreous cavity owing to difficulties in attaining an adequate drug concentration at the site following administration. More commonly, systemic administration or intraocular injection is used to deliver drugs to treat disorders associated with the vitreous cavity.23
TOPICAL ANTIBIOTICS
A factor that influences drug availability to the three anterior ocular target sites following topical ocular administration is retention of the antibiotic in the preocular area. The volume of tears in the eye is approximately 7 μL, most of which resides in the conjunctival sacs, with approximately 1 μL covering the cornea. The volume delivered by most commercial ophthalmic eye drop dispensers is approximately 30 to 50 μL. About one half of a typical drop is spilled from the eye almost immediately because the conjunctival cul-de-sac can contain only 20 to 30 μL.24 Reflex tearing and blinking induced by instillation increases spillage onto the skin, and causes drainage through the puncta to the nasolacrimal duct and subsequent absorption. Five minutes should elapse before instillation of a second eye drop, because reflex stimulation persists for approximately 5 minutes following instillation of an eye drop.25
Average tear production of 1 μL/min will continue to dilute agents in the tear film. The antibiotic must be present in the tear film in amounts above the MIC values of the pathogen for a significant amount of time to produce microbiological and clinical improvement or cure. When the target site is intraocular, drug must be absorbed from the preocular region into the eye. However, absorption of drug across the cornea is inefficient owing to its impermeable nature and small surface area.
For drug delivery, the cornea can be considered to be composed of three layers: the epithelium, stroma, and endothelium. Bowman’s layer and Descemet’s membrane are important anatomically but not significant rate-determining barriers23 Drugs penetrate the outer epithelium either by partitioning through the cells (intracellular) or by passing between the cells (paracellular or intercellular). Most drugs penetrate the cornea via the intracellular route, although the paracellular route predominates for hydrophilic drugs.21 It has been demonstrated that for lipophilic compounds, the epithelium offers little resistance to absorption and the major barrier is passage across the hydrophilic stroma and the endothelium. For hydrophobic compounds, the major barrier is the outer lipophilic epithelium, whereas the hydrophilic stroma offers little resistance to drug absorption.26
Most data on antibacterial concentrations achieved in the cornea are based on assay of the contents of “full-thickness” corneal buttons. In reality, a gradient is established across the cornea, with different concentrations of drug in the different layers of the cornea.27,28 Antibacterial concentrations at the site of infection of the cornea may, therefore, be either considerably higher than published corneal concentrations (if organisms are superficially located) or significantly lower.24
The corneal route has always been assumed to be the major route of entry into the eye; however, recent evidence suggests that penetration across the conjunctiva and sclera may also contribute significantly to penetration into the anterior chamber. Several studies have shown that the outer layer of the sclera has less barrier resistance to hydrophilic drugs than the epithelium of the cornea. More research must be conducted to determine the precise conjunctival/scleral route, the critical factors, and the importance of this pathway to drug absorption into the eye.29,30,31,32,33,34,35,36 Overall, the intraocular bioavailability of topical antibiotics is typically less than 10% of the topical dose (Fig. 5).36
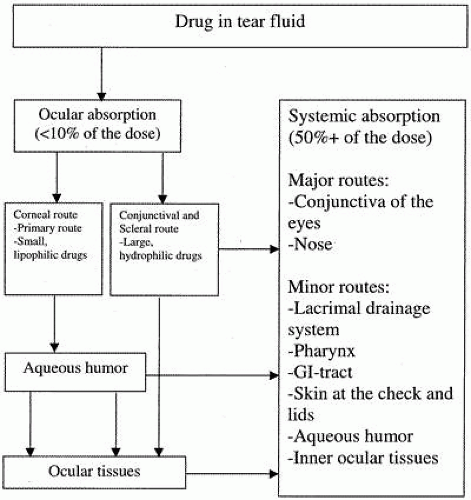 Fig. 5. Schematic diagram of ocular absorption. (Reprinted by permission from Worakul N, Robinson JR: Ocular pharmacokinetics/pharmacodynamics review. Eur J Pharmaceut Biopharmaceut 44:72, 1997.) |
Factors Affecting Absorption.
The solubility and pH of a topical antibiotic preparation are important factors affecting drug absorption. For example, when ciprofloxacin, more soluble at its commercial formulation of pH 4.5, comes into contact with tear film at neutral pH, its solubility equilibrium is shifted, and there is a tendency for the drug to precipitate.37 Increasing the concentration (but not the volume) of an antimicrobial eye drop will increase the level of drug in the precorneal tear film and, therefore, in ocular tissues. Hypertonic eyedrops dilute drug more rapidly than do less concentrated ones as a result of the osmotic effect through the conjunctiva38 Higher corneal concentrations of drug are achieved in abraded or inflamed corneas than in normal ones.39,40 Lipophilic antibiotics, such as chloramphenicol, penetrate the intact corneal epithelium more easily than do non-lipophilic antibiotics, such as gentamicin or cefazolin. However, with epithelium absent, as is the case with most infectious corneal ulcers, the epithelial barrier is eliminated, and hydrophilic antibiotics may enter the corneal stroma more easily.39,40 For instance, topical ciprofloxacin drops achieve anterior chamber levels 2 to 3 times greater if the epithelium is compromised than if it is intact.41,42
Distribution
Although precorneal and corneal factors discourage absorption, tissues in the anterior and posterior chambers are bathed continuously by circulating aqueous humor. Therefore, peripheral tissues are readily available for distribution, provided that drug properties, such as partitioning and binding, are optimal. Factors interfering with distribution include binding to melanin in the iris and the ciliary body, binding to protein in the aqueous humor, and rapid elimination of drug to an inactive site that can act as a biological reservoir, releasing drug slowly over time but producing relatively low therapeutic concentrations.21
Metabolism and Elimination
The time between drug instillation and its appearance in the aqueous humor is the lag time. Once drugs are absorbed into the anterior chamber, they are eliminated primarily by aqueous humor turnover, which is 1.5% per minute of the anterior chamber volume. When expressed as a half-life, aqueous turnover is 46.2 minutes, or 0.77 hours. Therefore, if drugs with a half-life of approximately 45 minutes (e.g., tobramycin) are eliminated from the eye, their elimination can be explained by aqueous turnover. For drugs with a half-life of less than 45 minutes, strong tissue binding is likely responsible for the longer half-life. If the half-life is less than 45 minutes, metabolism and uptake by blood vessels in the anterior uvea or iris are other likely pathways for elimination of drugs.21
Several antibacterials, including chloramphenicol and some fluoroquinolones, penetrate the cornea sufficiently rapidly to achieve potentially therapeutic concentrations in the aqueous humour if applied frequently. However, the constant flow of aqueous from the ciliary processes, where it is secreted, through the posterior and anterior chambers and out of the eye via Schlemm’s canal, prevents the accumulation of high concentrations of antibacterials at this site. Some absorption of antibacterials from the aqueous by the anterior uvea and lens is inevitable but not well characterized. Concentrations achieved in the vitreous body following topical administration are generally subtherapeutic, necessitating the administration of antibacterials systemically or by local (preferably intravitreal) injection for patients with infectious endophthalmitis.21
PERIOCULAR INJECTION
In general, hydrophilic drugs are more effective when given by the subconjunctival route. Absorption occurs from the reservoir of drugs at the conjunctival depot, which is not subject to precorneal factors as in topical application. Also, a subconjunctival drug bypasses the conjunctival epithelium, a significant rate-determining barrier for water-soluble drugs. The conjunctival route of administration often provides therapeutically effective drug levels for 8 to 12 hours after a single injection.21
In studies of subconjunctival injections of third-generation cephalosporins, the corneal levels achieved were fourfold higher than levels in aqueous concentrations. Concentrations in the choroid were fivefold to 15-fold higher than for the retina; retinal concentrations were about tenfold higher than vitreous cavity. Thus, there is a significant concentration gradient from choroid to retina and from retina to vitreous. In these studies, when the eyes were inflamed, the barriers appeared to be preserved. This report suggested that subconjunctival injections cannot replace intravitreal injections in the treatment of endophthalmitis.9,43 Similarly, subconjunctival injections are not as efficient as frequent topical application in treating keratitis and should be limited to situations where topical therapy is limited (eg, when the eye is patched or in uncooperative or noncompliant patients.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


