Ocular Pathology of Diabetes Mellitus
Peter F. Kador
Diabetes mellitus (DM) is a major health problem in the industrialized world and a rapidly escalating problem for developing countries, where urbanization is producing lifestyle changes that increase the risk of factors for the disease. By 2025, nearly 75% of all diabetic patients will be located in developing countries.1 DM is not a single disease but a group of metabolic disorders that are characterized by hyperglycemia with disturbances of carbohydrate, fat, and protein metabolism that are linked to altered insulin secretion, insulin action, or both.2 The etiologic classifications of DM recently have been changed to reflect the fact that hyperglycemia can result from different etiologic and pathophysiologic mechanisms.3 In addition, the diagnosis for DM has been simplified to two abnormal tests of either fasting plasma glucose levels of ≥126 mg/dL (7 mmol/L) (preferred), or 2-hour postprandial plasma glucose readings of ≥200 mg/dL (11.1 mmol/L), or a combination of both. Patients no longer are classified solely by insulin use, because, at some stage, all forms of diabetes may require insulin treatment. At present, DM is classified into four groups:
Type 1, formerly called insulin-dependent DM (IDDM), type I, or juvenile diabetes, is characterized by absolute insulin dependence resulting from the acute, autoimmune destruction of pancreatic β-cells.
Type 2, formerly called noninsulin-dependent DM (NIDDM), type II, or adult onset, is characterized by insulin resistance in peripheral tissues and defects in insulin secretion by the pancreatic β-cells. This is the most common form of DM. It is probably genetically based and linked to older age, obesity, and lack of exercise.
The third group has been classified as “other specific types,” in which diabetic diseases of various known etiologies are grouped together. This includes known genetic defects in β-cell function or insulin action, diseases of the pancreatic exocrine cells and their associated ducts, and dysfunctions associated with endocrinopathy, or caused by drugs, chemicals, or infections. Gestational diabetes is the fourth group. This group includes women who, during pregnancy, either develop type 1 DM or are discovered to have undiagnosed asymptomatic type 2 DM.
Central to the treatment of DM is the control of hyperglycemia. In cells with insulin-independent glucose transport, hyperglycemia initiates cellular changes that lead to the development of microvascular and macrovascular lesions. These lesions result in the onset and progression of the “late complications” of DM. These affect not only the eyes, but also the nervous system, cardiovascular system, and kidneys and result in vision loss, loss of limbs, neuropathy, and eventual death.
The importance of tight control of hyperglycemia in both type 1 and type 2 diabetic patients has been confirmed by the Diabetes Control and Complications Trials (DCCT) and the United Kingdom Prospective Diabetes Study (UKPDS) trials, respectively, both of which report that tight hyperglycemic control reduces the development and progression of diabetic complications.4,5 To define the specific role of glucose in diabetic complications, several biochemical mechanisms have been proposed. These include, in order of supportive evidence: (a) increased aldose reductase activity; (b) nonenzymatic glycation; (c) pseudohypoxia; and (d) diacylglycerol-protein kinase C pathway. Many of these mechanisms are strongly associated with oxidative stress and the presence of reactive oxygen species (ROS). The biochemical interplay between these mechanisms is summarized in Figure 18.1.
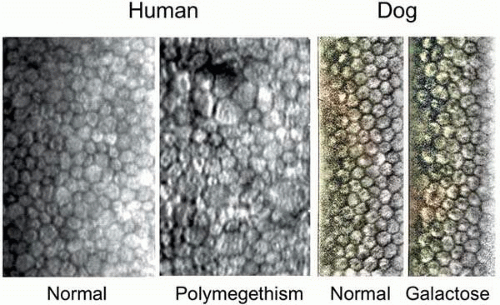 Figure 18.1. Interplay between hyperglycemia and glycolysis, polyol pathway, AGE generation, PKC activation, and ROS generation. AGEs, advanced glycation end products; Arg, arginine; Cit, citrulline; DAG, diacylglycerol; LDH, lactate dehydrogenase; DHAP, dihydroxyacetone phosphate; GP, glycerol-3-phosphate; GR, glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; NO, nitric oxide O2; oxygen; PA, phosphatidic acid; PKC, phosphokinase C; RAGE, receptors for advanced glycation end products; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle or citric acid cycle. |
Polyol Pathway
In most tissues with insulin-independent glucose uptake, glucose is rapidly phosphorylated by hexokinase and undergoes glycolysis.6 With hyperglycemia, hexokinase becomes saturated, and the increase in hypertonicity from excess glucose activates aldose reductase (AR) through gene expression. As a result, excess glucose enters the sorbitol pathway (polyol pathway), where it is converted to fructose. In the first step, glucose is reduced by AR and its cofactor nicotinamide adenine dinucleotide phosphate (NADPH) to the intermediate sugar alcohol sorbitol. In the second step, sorbitol is oxidized to fructose by sorbitol dehydrogenase (SDH) and its cofactor, NAD+. Unlike AR, SDH is not induced by hyperglycemia.7 As a result, glucose is reduced more quickly to sorbitol than sorbitol is oxidized is fructose. The net effect is the intracellular accumulation of sorbitol, a polar sugar alcohol that serves as an osmolyte.8 This excess formation of sorbitol has been linked directly to the onset and progression of diabetic complications. Support comes from a variety of pharmacologic studies utilizing lens and cell cultures, animal models, and aldose reductase inhibitors (ARIs). Moreover, the sorbitol pathway has been linked to downstream changes in phosphokinase C (PKC), vascular endothelial growth factor (VEGF), and redox potentials. Clinical support comes from recent observations that the severity of select diabetic complications is linked to the presence of select AR alleles.9,10 Specific evidence for ocular complications is discussed in subsequent sections of this chapter.
To investigate the specific role of AR, researchers have taken advantage of the broad substrate specificity of AR through the use of galactose-fed animals. Experimentally, galactose-induced tissue changes occur more quickly and progress to a more severe state than do glucose-induced changes.11,12 This is because (a) intracellular galactose is more rapidly reduced to galactitol by AR than glucose is reduced to sorbitol and (b) higher intracellular levels of polyol are achieved with galactitol, because it is not further metabolized by sorbitol dehydrogenase. Although not diabetic, the galactose-fed animal has become a specific model for investigating the role of AR in diabetes-like complications. If AR is involved in the mechanism(s) initiating the diabetic lesion, then the biochemical and pharmacologic results obtained with ARIs in the galactose-fed animals should be complementary to that in diabetic animals.
Although excess sorbitol formation resulting from increased AR activity appears to be the culprit, the specific mechanism(s) delineating how the polyol pathway contributes to the deleterious effects of hyperglycemia is still unclear. Extensive studies in the crystalline lens by Kinoshita suggest that neither sorbitol (nor galactitol) formation by itself, nor excess AR activity, specifically initiates the adverse biochemical effects associated with diabetic complications. Instead, the adverse effects are initiated by osmotic stress resulting from the intracellular accumulation of the sugar alcohols sorbitol or galactitol. Others propose that increased flux through AR, SDH, or both initiate oxidative stress through a redox imbalance linked to the excess utilization of NADPH or NADH.13 Oxidative stress generates ROS which, in turn, activates cell proliferation, apoptosis, tissue dysfunction, and cellular damage. Reduced NADPH levels from AR activity has been linked to diverse metabolic changes such as reduced levels of the antioxidant glutathione, the synthesis of nitric oxide (NO), and the activation of protein kinases.14 NADH utilization by SDH has been suggested to increase the synthesis of diacylglycerol (DAG) from dihydroxyacetone phosphate.15 Because SDH is inactive in galactose-fed animals, it has been suggested that osmotic stress occurs primarily with galactosemia, whereas ROS-generating oxidative stress occurs in diabetes. However, in animal studies, the administration of antioxidants and potent superoxide scavengers does not alter the effects of polyol pathway activity.16
Several reports and reviews also indicate that sorbitol is formed independently of AR, through the auto-oxidation of glucose.17 However, the auto-oxidation of glucose only occurs at high pH levels and not under physiologic conditions. In vitro studies at physiologic pH indicate that polyol formation only occurs when glucose or galactose are incubated in the presence of purified enzyme and adequate NADPH, not when the sugars are incubated only with NADPH.18,19 Transgenic and knockout mice also support the requirement for AR.20,21,22
Nonenzymatic Glycation
Glycation occurs when proteins or lipids containing free amino groups or ε-lysine nonenzymatically react with the aldehyde form of glucose to form a Schiff base through the Maillard or browning reaction. A well-known in vivo example is hemoglobin (HbA1c), the glycation of which correlates with hyperglycemia levels. The measurement of HbA1c is commonly used for retrospectively evaluating the long-term control of hyperglycemia in diabetic patients. The Schiff base then rearranges to an irreversible Amadori product, which further undergoes extensive rearrangement, degradation, and oxidation reactions to form advanced glycation end products (AGEs). This process is accompanied by the formation of free radicals and ROS.23 AGEs are chemically heterogeneous compounds. The structures for the majority of AGEs remain to be established. Because of their slow formation under physiologic conditions, AGEs primarily are associated with proteins possessing low turnover, such as lens proteins (crystallins) or extracellular matrix proteins such as collagen.24 AGE formation is accelerated with persistent hyperglycemia, dyslipidemia, and the oxidative stress of uncontrolled hyperglycemia, so that proteins with shorter turnover times, such as apolipoproteins and low-density lipoproteins (LDL), also can be modified.25
AGEs impact anatomic structural integrity through cross-linking, the direct generation of reactive oxygen intermediates, and the quenching of nitric oxide.26 In addition, AGEs can bind certain proteins called receptors of advanced glycation end products (RAGE). These proteins are members of the immunoglobulin superfamily of cell-surface molecules that are involved in signal transduction. RAGE are multispecific because they also are bound and activated by non-AGE molecules. It has not been established whether AGEs have a higher affinity and activating potential for RAGE, or whether many of these AGE-binding proteins are not accidental RAGE receptors.27 Nevertheless, experimental evidence, including murine models, indicate that RAGE are important in the development and progression of end-organ damage in diabetes.28,29,30,31 The relationship between AR activity and AGEs formation is complex. Fructose and its metabolites fructose-3-phosphate and 3-deoxyglucosone (3DG) are more potent glycating agents than is glucose. Therefore, increased fructose levels resulting from the activation of the polyol pathway should increase glycation. However, AR also reduces a number of potent glycating agents, such as methylglyoxal (MGO).32, 3-deoxyglucosone (3DG),33,34 and 4-hydroxynonenal (HNE),35 which are precursors of AGEs. In diabetic patients clinically administered the ARI epalrestat, HbA1c levels were not altered; however, plasma levels of Nε-(carboxymethyl)l ysine, a major epitope of AGE, were decreased significantly.36 Similarly, epalrestat reduced the levels of Nε-(carboxymethyl)lysine in erythrocytes.37 In diabetic patients undergoing hemodialysis, the clinical administration of epalrestat reduced the levels of 3-DG and AGEs, especially imidazolone, in erythrocytes.
Pseudohypoxia
The impaired mitochondrial oxidation of NADH in hypoxic tissues produces metabolic changes in vascular tissues that are similar to those observed with DM. These similarities have been linked to altered cytosolic NADH levels, which, in diabetic tissues, are postulated to result from increased flux through the polyol pathway where NAD+ is utilized by SDH in the conversion of sorbitol to fructose. Therefore, hyperglycemia induces a hypoxic state or pseudohypoxia in diabetic tissues in which the altered NAD/NADH levels result in the increased synthesis of diacylglycerol (DAG). This leads to inappropriate PKC activation, inhibition of fatty acid oxidation, increased prostaglandin synthesis, increased free radical production, and increased NO production.38 The suggestion that increased SDH activity, rather than AR activity, is critical to the formation of diabetic complications is controversial, having no firm data with SDH inhibitors established in ocular tissues. The importance of specifically targeting SDH inhibition also has been questioned, because ARIs indirectly control SDH activity by limiting the amounts of available sorbitol substrate. This concept does not explain why galactose-fed animals develop diabetes-like complications without the involvement of SDH.
Protein Kinase C
PKC is a member of the serine/threonine kinase family that regulates a variety of cell functions, including proliferation and differentiation, gene expression, cytoskeletal organization, cell migration, and apoptosis. PKCs catalyze the transfer of a phosphate group from ATP to various substrate proteins. PKC itself undergoes a series of complex phosphorylation steps before activation, during which it translocates from the cytosol to the membrane of the cell. A number of isoforms of PKC have been identified according to their structure and substrate specificities. These have been divided into least three subfamilies: conventional (α, βI, βII, γ); novel (δ, ε, θ, η); and atypical (λ, ζ, τ). PKC-μ, and PKC-ν possibly represent a distinct fourth subfamily, with little structural analogy to the other PKCs.39 High glucose levels stimulate the de novo diacylglycerol (DAG)-PKC pathway and subsequently stimulate ROS production through a PKC-dependent activation of NAD(P)H oxidase. Hyperglycemia induces several isoforms of PKC, including the PKC-β isoforms in the aorta and heart, and PKC-α, β, ε in the retina.40,41 PKC-α, β, δ, and ε are activated in the glomeruli of diabetic rats.42 The direct or chronic activation of PKC-ε plays a role in the generation of muscle insulin resistance by hyperglycemia.43
Because numerous PKC isoforms are present in tissues, but only a few are activated by hyperglycemia, the overall regulation of the entire PKC family could result in adverse biochemical effects. Moreover, not all hyperglycemia-induced activations of PKCs are necessarily adverse. For example, the activation of PKC-ε is essential for glucose transport and insulin resistance.44,45 Therefore, research has focused on the regulation of the β-isoforms, because increased activation of PKC-β in vascular tissues from diabetic animals and in vascular tissues exposed to elevated glucose have been linked to altered vascular blood flow, extracellular matrix deposition, basement membrane thickening, increased permeability, and neovascularization.46
Cornea and Conjunctiva
The anterior surface of the eye is covered by the corneal epithelium and conjunctiva. The limbus lies between. Optical clarity of the avascular cornea is critical for good vision. The clinical effects of uncontrolled DM on these tissues include decreased corneal sensitivity, increased corneal thickening, persistent stromal edema, and decreased tolerance to stress associated with photocoagulation, vitrectomy surgery, or even the wearing of contact lenses.47,48,49,50 Diabetic patients undergoing LASIK have poorer refractive results and increased risks of developing postoperative epithelial complications such as epithelial ingrowth.51,52 In addition, diabetes is a risk factor for corneal graft failure.53
Tear secretion is significantly reduced in diabetic patients, with conjunctival impression cytology showing a higher degree of squamous metaplasia and lower goblet-cell density. These changes appear to be related to metabolic control and the presence of diabetic neuropathy, and they correlate with the severity of diabetic retinopathy (DR).54 A positive correlation between increased HbA1c levels and reduced tear secretion in type 1 diabetes patients also has been reported.55
The corneal epithelium forms a barrier that separates the tear film from the inner corneal layers, and this barrier can be disrupted by noxious agents, trauma, or microbial infections.50 A continuous seal is formed by the tight cellular junctions (zonula occludens) located between the basal layer of epithelial cells just below the apical surface. These tight junctions regulate the paracellular pathway by which molecules move between adjacent epithelial cells.56 A number of tight junction-related proteins, including occludin, claudin-1, -2, -3, -4, -7, -9, and -14, and ZO-1, have been identified in human corneal epithelium.57 ZO-1 also has been observed in the midlevel epithelial cells of the rabbit cornea, where it is not associated with tight junctions but rather with a form of adherents junction. In this region, it is postulated that ZO-1 is associated with paxillin, a focal adhesion–associated phosphoprotein; together, the ZO1–paxillin adherents junctions function to reinforce attachments between the basal cells and wing-cell junctions.58 The adhesion of corneal epithelial cells also is regulated by plasminogen activator inhibitor-1 (PAI-1) and laminins.59
The corneal epithelial response to an immunologic assault or wounding involves a number of biochemical agents, including adhesion molecules, cytokines, chemokines, matrix metalloproteinases (MMP), and tissue inhibitors of metalloproteinases (TIMP).60 Corneal epithelial cells, through their production and release of cytokines such as interleukin-1 (IL-1), play a critical role in the initiation of ocular surface responses to potentially antigenic molecules entering corneal tissues.50 In addition, corneal epithelial cells actively participate in tissue remodeling via the degradation of the basement membrane by expression and activation of the MMP–TIMP system. The newly synthesized basement membrane components serve as a scaffold for epithelial cell migration. During wound healing, corneal epithelial cells also express various extracellular matrix (ECM) molecules that serve as a scaffold for the attachment and migration of the corneal epithelium. The expression of ECM is important in epithelial wound repair, because fibronectin derived from tears and/or secreted by keratocytes can mediate the binding of the epithelial cells to the denuded corneal surface via integrin receptors.
Diabetes-linked morphologic changes to the corneal epithelium include alterations in the size, shape, and number of epithelial cells, along with variations in the number of epithelial cell layers and areas of thinning.61 Diabetes also is associated with decreases in epithelial barrier function, with a 5.4-fold increase in fluorescein uptake reported.62 This increase correlates with the clinical severity of retinopathy as measured by the extent of retinal nonperfusion. Both corneal epithelial cell morphology and barrier function are regulated by the MAP kinase signaling pathway. Its activation results in decreased barrier function.63 Activation of PKC, which occurs with hyperglycemia, also activates MAP kinase, and this results in the disruption of tight junctions within the corneal epithelium.64 In the corneal epithelial basement membranes, diabetes-linked changes include membrane thickening and multilayering. Decreases in the membrane components, α3-integrin and β1-integrin, and increased expression of matrix metalloproteinase-10 has been reported, whereas stromal ECM components remain unchanged in diabetic corneas.65 These alterations may be linked to either increased degradation or decreased synthesis of basement membrane components and related integrins, and they may directly contribute to the reduced epithelial adhesion and other wound-healing abnormalities found in diabetic corneas.66 With high glucose concentrations, a loss of homeostatic levels of laminin-5 occurs, and this has been postulated to be linked to clinically observed weakened epithelial cell adhesions.67
Stromal thickening in diabetic corneas has been reported. Analysis of corneal buttons from diabetic and nondiabetic animals indicates that this thickening is due primarily to hydration.68 An increase in the autofluorescence of the stroma also has been reported, and this is linked to the presence of morphologic changes in the corneal endothelial cells.61,69
Increased wrinkling of the Descemet membrane—the basement membrane of corneal endothelial cells—has been reported with DM.70 This wrinkling is thought to be secondary to increased corneal hydration.71 No significant basement membrane thickening has been reported;72 however, an increase in abnormal posteriorly located banded material similar to that observed with Fuchs dystrophy and aphakic bullous keratopathy has been reported.73,74
Corneal endothelium is the single layer of cells forming a boundary between the corneal stroma and anterior chamber. The barrier and “pump” functions of the endothelium are responsible for maintaining corneal transparency by regulating stromal hydration. In the diabetic cornea, endothelial cells demonstrate a loss of hexagonality, size (polymegathism), and shape (pleomorphism), with changes in cell area being the most discriminating morphologic variable (Fig. 18.2).61,75,76 These changes reflect endothelial loss that makes the cornea more susceptible to stress-related injury. Markedly irregular F-actin fibers crossing the endothelial cell cytoplasm have been reported in diabetic corneas.77 These abnormal F-actin patterns have been proposed to contribute to the polymegathism observed in diabetic corneal endothelial cells, and they may be a result of constant stress in the regulation of cell volume.77
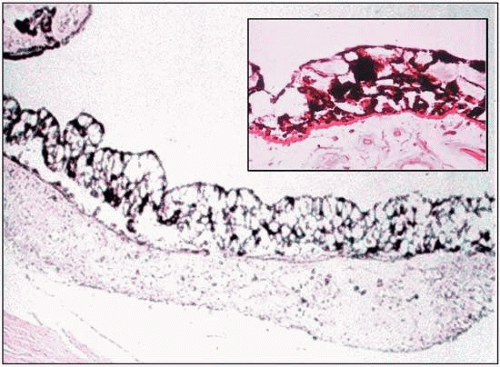 Figure 18.2. Specular microscopy of human and dog corneal endothelial cells. Polymegathism, commonly observed in diabetics, is dramatically demonstrated on the right human photograph. The corneal endothelium from the galactose-fed dog has similar but less dramatic changes in size, along with loss of hexagonality. Pictures were photo-enhanced for better visualization of the cells. |
Aldose Reductase and Corneal Changes
AR has been demonstrated by immunohistochemistry to be present in the corneal epithelium and endothelium.78 Studies conducted with a number of structurally diverse ARIs in diabetic and galactose-fed rats and dogs, and clinically in humans with DM, demonstrate that ARIs are effective in ameliorating changes in both corneal epithelium and endothelium structure and function and in neuropathy of the cornea. Following clinical observations during the 1970s that diabetic patients undergoing vitrectomy often had corneal resurfacing and wound-healing complications, Kinoshita and associates demonstrated that similar corneal problems can occur in rats. Delays in the rate of re-epithelialization and the hazy, edematous appearance of the healed corneas were documented in both severely diabetic and galactose-fed rats whose corneal epithelium had been totally denuded.69,79 Histologic studies indicated that the epithelial cells had a distended appearance and an absence of filopodia, suggesting that the corneal epithelial cells are osmotically altered. Both the delay in the rate of re-epithelialization and the edematous appearance of the healed cornea were corrected by the concomitant oral or topical administration of ARIs such as sorbinil.
Subsequently, these observations were expanded to include demonstrations that a number of structurally diverse ARIs, including sorbinil, M79175, CT-112, AL 1576, and SNK 380, administered either topically or orally not only normalized wound healing, but prevented corneal sugar alcohol accumulation and reduced epithelial permeability and loss of nerve sensitivity in the corneal epithelium. Corneal thickening and persistent stromal edema suggestive of abnormal endothelial cell function, as well as morphometric changes in cell size and the loss of hexagonality, also were ameliorated.80,81,82,83,84,85,86,87 These changes were confirmed by histologic and ultrastructural techniques, as well as by specular microscopy. Corneal sensitivity was measured using a Cochet-Bonnet aesthesiometer, and the nerve changes were documented by ultrastructural studies that indicated that the morphologic changes associated with the degeneration of axons and mitochondria were inhibited by the ARIs.
Specular microscopy of diabetic and galactose-fed dogs also indicates morphologic changes in the size and shape of endothelial cells (see Fig. 18.2).88,89 In galactose-fed dogs, these morphologic changes were reduced by the oral administration of sorbinil or M79175;88 however, these corneal changes were not reversible after 24 months of galactose feeding, when retinal changes had clinically progressed to the background retinopathy stage.90 In the nonoperated eyes of galactose-fed dogs, corneal barrier function measured using anterior fluorophotometry also was maintained by the oral administration of epalrestat (Kinedak).91
A number of reports describe the clinical use of select ARIs. These began with findings that the topical application of sorbinil promoted corneal healing in diabetic patients with corneal epithelial disorders unresponsive to conventional treatment.92 These findings were subsequently confirmed with the use of topical CT-112 in two diabetic patients—one with recurrent corneal erosion after vitrectomy and the other with spontaneous superficial punctate keratopathy. Clearing was observed within 2 months of treatment, but discontinuing CT-112 resulted in the reappearance of the corneal lesions. These changes again disappeared when drug was resumed.93 In addition to healing corneal erosion, the 3-month oral administration of epalrestat to 15 aphakic or pseudophakic diabetic patients resulted in the significant recovery of corneal sensation, reduced rose bengal and fluorescein staining of the corneal epithelium, and improved tear production and prolonged tear break-up time.94 However, in another 12-week study of 12 pseudophakic diabetic patients treated with oral epalrestat, significant improvement was observed only in fluorescein staining scores and conjunctival sensation. Minor improvement in rose bengal staining, tear clearance, and corneal sensation, but no improvement in tear production, tear break-up, or morphologic changes in corneal epithelium and endothelium, was observed.95 The topical application of CT-112 to 18 diabetic patients significantly improved corneal sensitivity and reversed abnormal epithelial cell morphology, compared with 18 similar diabetic patients receiving placebo.96 Topical CT-112 also improved corneal epithelial barrier function in 22 diabetic patients, compared with 12 placebo-treated diabetic patients.97 Significant improvement was observed within 4 weeks of treatment. In the corneal endothelium, morphologic variations in polymegathism and pleomorphism were reduced in eight diabetic patients treated for 12 weeks with topical CT-112, compared with five similar diabetic patients receiving placebo.98
Nonenzymatic Glycation and Corneal Changes
The role of nonenzymatic glycation on diabetes-related corneal changes has not been extensively investigated, and no extensive animal studies have been conducted. Several clinical reports indicate that corneal autofluorescence is increased in diabetic patients. Corneal AGE fluorescence has been found to be significantly higher in type 2 diabetic patients with proliferative DR, compared with diabetic patients without such retinopathy.99 Similarly, a 1.6-fold increase in autofluorescence has been reported in type 2 diabetic patients. However, the observed increase correlated with age rather than with blood glucose levels. Corneal autofluorescence is linked not only to AGE formation, but also to reduced pyridine nucleotide levels.100 Because the cornea is avascular, corneal autofluorescence related to AGEs is believed to be formed by glucose that reaches the cornea via the aqueous humor. However, autofluorescence was not observed to increase in diabetic patients with increased blood–aqueous barrier permeability.101 These results suggest that the autofluorescence attributed to AGEs in the cornea may be due to factors other than hyperglycemia or increased aqueous glucose levels, or even the nonenzymatic glycation proteins. Immunoreactive N(ε)-carboxymethyl lysine (CML) has been reported to be selectively present in the epithelial basement membrane of diabetic corneas. In vitro, nonenzymatic glycation of laminin on culture dishes attenuated adhesion and the spreading of corneal epithelial cells. Addition of the glycation inhibitor aminoguanidine to the incubation mixture inhibited CML formation and promoted the adhesion and spreading of corneal epithelial cells in a dose-dependent manner.102 No in vivo studies on the effect of aminoguanidine on corneal changes in diabetic animals have been conducted.
An interesting potential link between nonenzymatic glycation and dry eye recently has been reported in diabetic rats.103 Compared with nondiabetic rats, significant decreases in lacrimal gland weight and tear film volume were observed in diabetic rats, along with elevated tear levels of the proinflammatory cytokines IL1B and tumor necrosis factor (TNF). Expression of AGE, RAGE, and NFκB was also increased in their lacrimal glands. The increased presence of proinflammatory cytokines in tears, and the higher lacrimal gland expression of AGE, RAGE, and NFκB, suggest that these factors are involved in signaling and possibly in the inflammatory changes that can lead to dry eye.
PKC and Corneal Changes
Even less is known of the relationship between PKC and diabetes-induced corneal changes. In nondiabetic corneas, transparency is linked primarily to the architectural arrangement of stromal collagen, which contains type I collagen. The maintenance of stromal integrity is believed to be due to a coordinated balance of collagen synthesis and degradation.50,104 PKC signaling is believed to play an important role in corneal collagen degradation.105 Transparency also is lost with neovascularization, which involves the sprouting of new vessels, essentially from capillaries and venules of the pericorneal plexus. VEGF is up-regulated in inflamed and vascularized corneas in humans and in animal models.106 Activation of PKC is a major signaling pathway required for VEGF-induced proliferation and angiogenesis.107 PKC activity in diabetic corneal tissues has not been reported; however, hyperglycemic activation of PKC requires AR.108 Because PKC is downstream of AR, PKC activity is reduced by ARIs.
Iris
In the diabetic iris, lacy vacuolization of the pigment epithelium is present in approximately 40% of eyes (Fig. 18.3).109,110 These vacuoles contain glycogen.111 The cause of this glycogen storage is unknown; however, it has been suggested to be linked to blood sugar levels.112 In addition, an increase in microcysts of the iris pigment epithelium has been described in association with DM.113
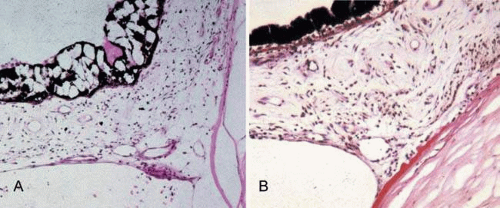 Figure 18.3. Hematoxylin and eosin (H&E)-stained section of diabetic iris illustrating diabetic lacy vacuolization. Large vacuoles are present in both the anterior and posterior layers of the pigmented epithelium. Insert is a PAS-stained section indicating that the vacuoles contain periodic acid-Schiff-hematoxylin (PAS)-positive material. (Courtesy of Gerald R. Christensen, University of Nebraska Medical Center.) |
During intraocular surgery, as the intraocular pressure (IOP) is suddenly reduced with the opening of the anterior chamber, the glycogen-containing vacuoles of the diabetic iris pigment epithelium are prone to rupture. This can result in a dispersion of melanin pigment that then can be phagocytosed by the corneal endothelium cells.114 Dilation of diabetic pupils also can result in the release of pigment into the anterior chamber.115
Pinpoint holes in the iris, which are clinically observed by slit lamp using transpupillary retroillumination, also are increased in diabetic patients. They are more prevalent in light-colored irises, with blue irises having the highest incidence. In type 2 diabetic patients, a 27% increase in the incidence of iris transluminance has been reported. The formation of these pinpoint holes is associated with the presence of severe, recent onset retinopathy rather than IOP or glycemic control. These observations have led to the suggestion that the formation of these defects is linked to hypoxia.116
Diabetic patients demonstrate pupillary abnormalities that are associated with autonomic nervous dysfunction. These include reduced pupillary response to light and insufficient dilation of pupils in response to darkness or mydriatics.117,118,119 The size of the pupil is linked to a balance between the input of the sympathetic and parasympathetic systems, with the common dysfunction being an exaggerated miosis due to failure of the sympathetic tone to counteract pupillary constriction by the parasympathetic system. Damage to the pupillary parasympathetic supply has been reported to occur in diabetic patients before the pupillary sympathetic pathway is affected.120 Sympathetic denervation also has been demonstrated in diabetic patients with pre- and postganglionic dysfunction of the sympathetic plexus.121 In diabetic children, insufficient pupil dilation in response to darkness is associated with a longer duration of DM and poor glycemic control.122
Ultrastructural studies suggest that these abnormalities are linked to changes in both the iris muscle cells and nerves.123 Changes observed by electron microscopy in the muscle cells of diabetic patients include the cytoplasmic presence of vacuoles and concentric lamellar membranous structures. Although present in both dilator and sphincter muscles, the dilator muscles appear more affected. Degenerative nerve endings containing lamellar membranous structures and diminished nerve fibers also were observed. In addition, the diabetic irises contained increased lipid droplets associated with pigment granules. All these changes increased with the duration of DM and poor glycemic control. Similar studies of iris muscles and innervating nerves obtained from cataract surgery indicate changes at the nerve terminal innervating the dilator muscle but not the sphincter muscle in patients with DM. These changes included the presence of mitochondrial abnormalities, dense bodies, and lamellar structures. Both diabetic and nondiabetic subjects had moderate muscle changes, especially in the sphincter, and these were attributed to aging.124 Glycogen levels also may contribute to dilator muscle damage.124
Neovascularization of the iris in diabetic patients is never primary, but always secondary to other ocular disorders. Neovascularization of the iris, clinically called rubeosis iridis because of the reddish coloration imparted by the new vessels on the iris surface, generally is associated with proliferative retinopathy. It is present in nearly two-thirds of all diabetic patients with proliferative retinopathy. Neovascularization of the iris anterior surface can start at either the anterior chamber angle, the pupillary border, the area in between the angle and pupillary border, or all three.125 Although fine new vessels clinically appear on the iris surface, they actually extend from and penetrate the iris stroma and appear in all layers. New vessel growth eventually progresses over the anterior chamber angle (Fig. 18.4). Infrequently, the process can remain stationary for years, with the anterior chamber angle remaining open without angle rubeosis. When neovascularization (rubeosis) of the chamber angle occurs, the new vessels cause a secondary glaucoma. However, as the connective tissue framework of the newly growing vessels adheres to the cornea and iris, a peripheral anterior synechia is formed that can rapidly close off the trabecular meshwork and give rise to chronic secondary angle-closure glaucoma (neovascular glaucoma). The new vessels and associated fibrous tissues can proliferate across the pupil and sequester it. Contraction of the fibrovascular tissue on the anterior iris surface also can occur. This causes traction of the pupillary margin and results in the structure being turned outward to form an ectropion uveae.
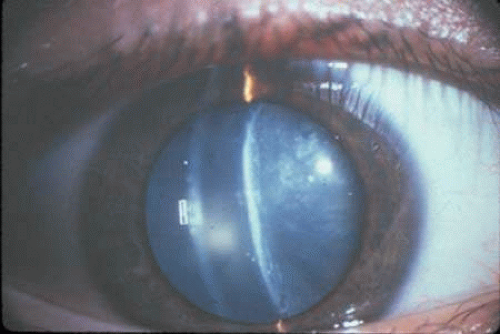 Figure 18.4. A. Severe vacuolization of the iris pigment epithelium and lush neovascularization, with abundant endothelial cells from a 36-year-old man with type 1 DM. B. Extensive anterior synechia with a fibrovascular growth extending from the Descemet membrane onto the surface of the iris in a 66-year-old woman with DM and absolute glaucoma with no light perception (PAS). (Courtesy of National Eye Institute David Cogan Collection.) |
The new vessels are thin-walled, have increased permeability, and can bleed easily. They have a tendency to be located toward the surface of the iris, but they can be anywhere within the stroma of the iris. A thin, transparent layer of myofibroblastic proliferation anterior to the new vessels also has been observed by scanning electron microscopy. The myofibroblasts have contractile properties and are proposed to be responsible for the angle closure of the neovascular glaucoma.
Studies associate VEGF with neovascularization of the iris, and this neovascularization has been proposed to be initiated by VEGF produced in the retina.126 AGEs have been reported to increase VEGF mRNA levels in the ganglion, inner nuclear, and retinal pigment epithelial (RPE) cell layers of the rat, and in vitro AGEs increased VEGF mRNA and secreted protein in human RPE and bovine vascular smooth muscle cells.126 Experimentally, intravitreal injection of the angiogenic peptide VEGF induces iris neovascularization in cynomolgus monkey eyes (Macaca fascicularis), with the formation of dilated and tortuous iris vessels that leak fluorescein.127 The intravitreal injection of an antisense oligodeoxynucleotide against VEGF reduced the level of iris neovascularization in monkeys.128 Iris neovascularization induced by the occlusion of three major retinal branch veins in cynomolgus monkeys was observed to occur in three phases.129 In the early phase, vessel dilation and intense uptake of thymidine into the vascular endothelial cells was observed. In the second, intermediate phase, thymidine uptake into the endothelial cells was reduced, but uptake into the iris stromal cells increased, along with the formation of a neovascular membrane in association with the anterior migration of stromal cells. During this phase, prominent new vessels, ectropion uveae, peripheral anterior synechiae, and elevated IOP developed. The third and final phase was characterized by a marked reduction of thymidine uptake and the regression of the neovascular membrane.
Role of Aldose Reductase in Iris
Mounting clinical and experimental evidence suggests that ARIs are associated with changes in the diabetic iris. The clinical administration of the ARI epalrestat has beneficial effects on pupillary light reflex associated with diabetic neuropathy, with significant improvement in the minimum diameter after light stimuli, constriction ratio, and maximum velocity of constriction observed.130 In contrast, no effect was observed in a small trial of 15 patients with the ARI ponalrestat (Statil) on diabetic autonomic neuropathy evaluated by cardiovascular reflexes and pupil reflex.131 However, ponalrestat also has been reported to be ineffective on other neuropathies due to its inability to penetrate human nerves.132 In type 1 adolescent diabetic patients, the presence of the Z-2/Z-2 genotype of AR, which results in increased AR activity, has been linked to a threefold increased risk of pupillary abnormalities that are an early indicator of diabetic autonomic neuropathy.133
Animal studies also demonstrate that ARIs are involved in diabetic iris pathophysiology. AR is present in the iris pigment epithelial cells of nondiabetic rats. It is unknown whether AR also is present in iris vessels because immunohistochemical detection of the enzyme in trypsin-digested iris vessels has not been successful, presumably due to the high amounts of collagen present. In the absence of synechiae, the irises of rats fed a 50% galactose diet display both a delay in dilation and a failure to fully dilate in response to treatment with mydriatics.134,135 In these rats, an initial delay in dilation, which is accompanied by a hypersensitivity to phenylephrine, is followed by the formation of fibrous tissue containing collagen, fibroblasts, and macrophages. This formation progresses beneath the posterior surface of the iris from the pupillary margin toward the iris root. Increased iridal blood vessel permeability to horseradish peroxidase (HRP) and 125I-albumin also has been observed.134,135,136,137,138 All these iridal changes, which include the delay in dilation, fibrous tissue formation, and altered vessel permeability, are absent in galactose-fed rats concomitantly treated with ARIs.
A disruption of the iridal blood–aqueous barrier has been reported in streptozotocin-diabetic rats, using light and electron microscopy with HRP.139 In rats diabetic for 2 to 6 months, HRP-labeled vesicles were observed in those iris endothelial cells that opened into the basement membrane and in intercellular junctions of the endothelial cells. HRP also was found consistently in the stroma around the vessels, and it seemed to reach the anterior chamber through the intercellular spaces between the fibroblasts. The passage of HRP was blocked by the posterior epithelial cells. Disruption of the blood–aqueous barrier also has been observed in galactose-fed rats.140,141 Electron microscopic examinations of irises from 14-month galactose-fed rats indicated increased leak-out of HRP around the iris vessels, with HRP observed to be present inside endothelial cell vesicles and in some endothelial cell junctions. A similar increase in permeability to Evans blue dye, which irreversibly binds to albumin to form a dye-protein complex, also was observed. This permeability to both HRP and Evans blue was prevented or reduced by treatment with ARIs. In addition to increased permeability, changes in the diameter of iris vessels, characterized mainly by a reduced vessel lumen (and apparent reduced blood flow), decreased iris vessel tortuosity (focal straightening), and neovascularization, also were observed. These structural changes, which correlate with chronic iris ischemia, also were prevented or reduced by treatment with ARIs. In contrast, ARI could not normalize blood–aqueous barrier permeability in Lewis rats, in which permeability was increased by uveitis induced through immunization with bovine retinal soluble antigen. This supports the premise that AR activity is linked only to hyperglycemia-associated iris blood aqueous permeability.
Lens
Diabetes is associated with physical, refractive, and cataractous changes of the lens. Clear lenses from diabetic patients are larger, with a widened subcapsular clear zone, when compared with lenses from age-matched nondiabetic subjects.142 Diabetic lenses appear to be approximately 15 years older than their age-matched counterparts and have increased levels of fluorescence. The fluorescence is linked to increased protein glycation in the cortex and nucleus.143 Lens fluorescence appears to be proportional to glycemic control, with increased lens fluorescence and decreased lens transmittance delayed by good metabolic control.144,145 Therefore, lens fluorescence provides information about the long-term control of diabetes.145
Hyperglycemia is also the major cause of transient refractive changes in diabetic patients.146 In general, hyperglycemia results in myopia, whereas a decline in blood sugar levels in response to appropriate therapy can result in hyperopia. However, hyperglycemic hyperopia or changes in both also have been reported.147 Intralenticular pressure changes result from the accumulation of glucose and its metabolic products, which include the osmolyte, sorbitol.148 As the intralenticular concentrations of these sugars increase, an osmotic gradient is formed that results in lens hydration and subsequent lens swelling. This swelling can be accentuated by rapid decreases in blood and aqueous glucose levels that can magnify the osmotic differences between the lens and aqueous and lead to an additional accumulation of water. If severe enough, these refractive changes result in lens opacification (i.e., cataract formation).
Several types of lenticular opacities develop in this diverse clinical population, which includes both juvenile and adult-onset diabetic patients. The most common diabetic cataracts contain cortical and/or posterior subcapsular opacities (Fig. 18.5). These readily form in type 1 (juvenile) diabetic patients and are characterized by bilateral snowflake-type cortical deposits and posterior subcapsular cataract.149 These opacities often are referred to as “true” diabetic cataracts, because they rapidly evolve over a period of days and generally appear simultaneously in both eyes. Their development appears to depend on the severity and prolonged poor control of the disease, rather than on the duration of the DM.150
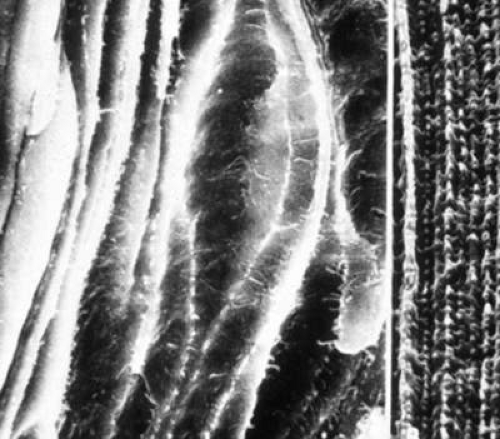 Figure 18.5. Slit lamp image of diabetic cataract from a 15-year-old girl with type 1 DM showing early cortical opacities with water clefts and clouding of anterior and posterior cortical lens fibers. (Courtesy of Manuel Datiles, National Eye Institute.) |
After the fourth decade of life, diabetic cataracts are generally difficult to differentiate from those of nondiabetic subjects.150 Nevertheless, a number of studies indicate that cortical and/or posterior subcapsular opacities are still prevalent in type 2 diabetic patients.151,152,153,154,155,156 Moreover, in the Barbados Study, the presence of cortical and posterior subcapsular opacities positively correlated with glycated hemoglobin levels.157 Adult-onset diabetic cataracts also often contain nuclear opacities that closely resemble the typical senile cataracts of nondiabetic subjects.158 Nevertheless, type 2 diabetic patients, especially those with poor control, undergo cataract extraction at a higher rate than do nondiabetic patients of comparable age.
Only limited ultrastructural studies have been reported on human diabetic lenses. The epithelial cell density has been observed to decrease in mature-onset diabetic lenses.159 Examination of the cataractous areas from mature-onset diabetic lenses and senile posterior subcapsular cataracts revealed no difference between the two.160 Both cataractous regions contained similar spherical globules up to 20 mμ in diameter, with the area of globular degeneration usually 300 mμ deep, but with deeper fusiform extensions. Immunofluorescence suggested that the globules contained primarily γ-crystallin, along with α– and β-crystallin and actin. They also contained higher concentrations of cysteine or cystine than the surrounding lens tissue, but they did not react to stains for carbohydrate or calcium. Light scattering from the globules was estimated to account for the majority of opacity present, with light scatter from fiber degeneration accounting for the remainder.
A similar examination of lens nuclei from late-onset diabetic patients using transmission electron microscopy revealed extensive cellular damage to the exterior region of the diabetic lens nuclei, especially at the cortical–nuclear interface (161). Lens fiber condensations, as well as areas of cytoplasmic loss, were observed in the outer areas of the nucleus along with multilamellar membrane aggregates, voids where cytoplasmic material was lost, extracellular space deposits, density variations between adjacent fiber cells, and heterogeneously staining globules. Little cell damage was observed in the opaque central region of the nuclei except for the irregular shape and packing of the fiber cells. However, the ultrastructure of inner nuclear fiber cells did not differ from that seen in the normal lens and in lenses from nondiabetic age-related nuclear cataracts.
Closely related to diabetic cataracts are sugar cataracts, resulting from abnormal galactose metabolism caused by the absence of either galactokinase, UDP-galactose transferase, or UDP-galactose epimerase.162 All result in increased levels of galactose and the subsequent formation of the sugar alcohol, galactitol. In young children, uncontrolled galactosemia leads to bilateral cataracts within several weeks. Total or partial galactokinase deficiency combined with high milk consumption may be linked to periods of increased hypergalactosemia and accelerated senile cataract formation.163 This premise is supported by studies of Asian populations but not of Italian.164,165
Aldose Reductase and Sugar Cataracts
Animal studies have elucidated the common mechanism by which sugar cataracts associated with diabetes and galactosemia develop.166 From initial histopathologic findings that lenses from diabetic and galactosemic rats display similar hydropic lens fiber cells that eventually rupture, with liquefaction of the ruptured cells appearing as vacuoles (Fig. 18.6), and the observation by van Heyningen that AR is present in the lens, Kinoshita demonstrated that the AR-catalyzed intracellular accumulation of the sugar alcohols sorbitol and galactitol initiates osmotic changes that lead to sugar cataract formation.167 Therefore, the common pathway of sugar cataract formation involves polyol formation, lens swelling, membrane permeation, vacuole and cleft formation, disturbance to the intracellular environment, protein aggregation or modification, and light scatter. This “osmotic hypothesis” has been supported by a variety of studies utilizing lens culture, animal models, and ARIs.167,168,169,170,171,172,173,174,175 Moreover, animal studies show that the onset and severity of sugar cataract formation is linked directly to the AR-catalyzed accumulation of polyols.21,176,177 Cataracts easily develop in the rat and South American degus (octodon), in which lens AR levels are high, but not in the mouse, in which AR levels are low. Sugar cataracts, however, do form under both diabetic and galactosemic conditions in transgenic mice expressing rat lens AR in their lenses.21 Similarly, despite a nearly equal prevalence of diabetes in cats and dogs, sugar cataracts are not commonly observed in cats compared to dogs because the lenticular AR activity is much higher in dogs than in age-matched cats.178,179 If diabetic cataract formation was independent of AR, then an equal incidence of cataracts in both cats and dogs would be anticipated, because the incidence of diabetes and levels of hyperglycemia in cats and dogs are similar. In addition to osmotic stress, recent studies also indicate that oxidative stress is induced by the formation of sorbitol or galactitol.22,180,181,182,183 Both osmotic and oxidative stress is prevented through the inhibition of polyol formation.
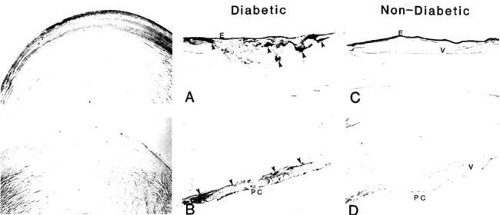 Figure 18.6. Electron micrograph of lens fibers from a normal lens (right) showing normal interdigitations of lens fibers and diabetic lens (left) illustrating swollen and ruptured fibers. Note loss of interdigitations. (Courtesy of Toichi Kuwabara, National Eye Institute.) |
Sorbitol Dehydrogenase Inhibition and Cataracts
Sorbitol dehydrogenase inhibition also supports the importance of sorbitol formation in the lens. In contrast to the concept that inhibition of sorbitol dehydrogenase ameliorates oxidative stress that can lead to hypoxia, inhibition of sorbitol dehydrogenase actually enhances the formation of diabetic cataracts. This was observed both by treating diabetic rats with a sorbitol dehydrogenase inhibitor184 and by breeding diabetic transgenic mice containing AR in their lens with mice deficient in lens sorbitol dehydrogenase.21
Nonenzymatic Glycation and Sugar Cataracts
Hyperglycemia also is linked to nonenzymatic glycation and oxidative stress, and it has been suggested that these are the cause of sugar cataracts.150,185,186 Clearly, whereas diabetic patients with increased levels of AGEs also have a significantly higher proportion of HMW proteins, 187 biochemical studies and several clinical trials with antioxidants do not confirm their importance in cataracts.188,189,190 Moreover, if sugar cataract formation was independent of AR activity and initiated by glycation and oxidative stress, then one would expect cataracts to form in both hyperglycemic mice and cats to the same extent as they do in rats and dogs. Furthermore, ARIs inhibit sugar cataracts in both diabetic and galactosemic animals without reducing the levels of either hyperglycemia or galactosemia, lens protein glycation, or AGE formation.191,192,193 This indicates that the inhibition of ARIs is the primary key to controlling sugar cataract formation.
Aldose Reductase and Human Sugar Cataracts
Osmotic changes are well documented in the sugar cataract in infants and numerous cases of acute diabetic cataract formation have been reported in diabetic patients ranging from adolescents to adults.194,195,196,197,198,199,200 These acute cataracts rapidly form within hours to days and are generally associated with the treatment of acute hyperglycemia. However, many question the importance of osmotic stress in the formation of adult sugar cataracts because AR activity in the adult human lens is significantly lower than that in rodents. Moreover, human sugar cataracts differ in appearance, with predominant cortical and posterior subcapsular opacities formed, in comparison with the anterior cortical involvement in rodents. Nevertheless, the presence of select alleles of the AR gene have been linked to the clinical development of diabetic cataracts in adults.201 Both sorbitol and galactitol accumulate in in vitro cultured human lenses, and sorbitol accumulation is higher in lenses from diabetic patients compared with those from nondiabetic subjects. This accumulation is inhibited by the presence of ARI.202 Moreover, the levels of sorbitol and fructose recovered from cataracts extracted from diabetic patients parallels HbA1c and fasting blood sugars levels.203,204 The immunohistochemical distribution of AR in the human lens also parallels that in the adult rat, with staining in both limited to the epithelium and superficial cortical fibers.78,205 Immunohistochemical staining for AR in the superficial cortical layers of rat lenses increases after the induction of diabetes or galactosemia.206 Similarly increased immunoreactive staining for AR also has been observed in both the anterior or posterior superficial cortical layers of cataractous lenses extracted from diabetic versus nondiabetic subjects (Fig. 18.7).206 This staining mirrors the localized lens hydration observed by Raman spectroscopy, which primarily appears in the cortical region and not in the nucleus.207
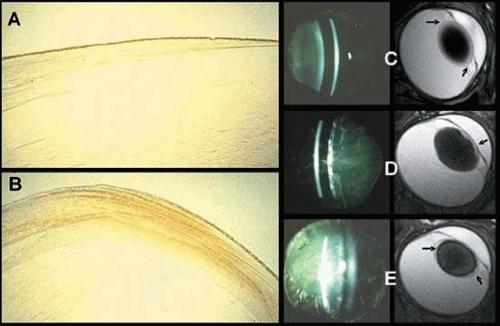 Figure 18.7. Aldose reductase in the human lens. The top left panel shows the distribution of AR in the human lens treated with antibodies against human placental AR and visualized with 3,3′-diaminobenzidine and hydrogen peroxide method (DAB-PAP). AR staining was observed primarily in the epithelium and the superficial cortical fibers. The bottom left panel represents the stain control only treated with HRP-DAB but not AR antibody. The center and left panels show immunostained anterior (A, C) and posterior (B, D) portions of surgically removed cataractous lenses. Increased AR staining was observed in both the anterior (A) and posterior (B) cortex of the lens from patients with DM. No increased staining was observed from cataractous lenses from age-matched nondiabetic patients (C, D). E, epithelium; PC, posterior capsule; v, vacuoles; arrows show AR staining associated with vacuoles. |
Some of the observed differences between rat and man may be clarified by studies of sugar cataracts conducted in dogs, in which the specific activity of lens AR is similar to that in man.208 The immunohistochemical distribution of AR in dog lens is also similar to that in the rat and human (Fig. 18.8).78,205,209 Clinically, the early stages of sugar cataracts in diabetic and galactosemic dogs is similar,208 and sugar cataract formation has been reduced in galactose-fed dogs in a dose-dependent manner with ARIs (Fig. 18.9).209,210 Extensive studies in galactose-fed dogs indicate that the onset and severity of sugar cataracts is age-dependent and that this mirrors the age-dependent activity of AR in the lens.176 Sugar cataracts begin with an accentuation of both the anterior and posterior suture as a result of lens fiber swelling.208,209 This is followed by equatorial vacuole formation and eventually the appearance of opacities throughout the anterior and posterior cortex. The cortical opacities may be followed by the appearance of hypermature cataracts with nuclear involvement; however, in older dogs, the progression usually is limited to cortical opacities with apparent peripheral cortical clearing. In galactose-fed dogs less than 6 months of age, dense, mature cataracts rapidly form; this is followed by rapid lens resorption and uveitis.
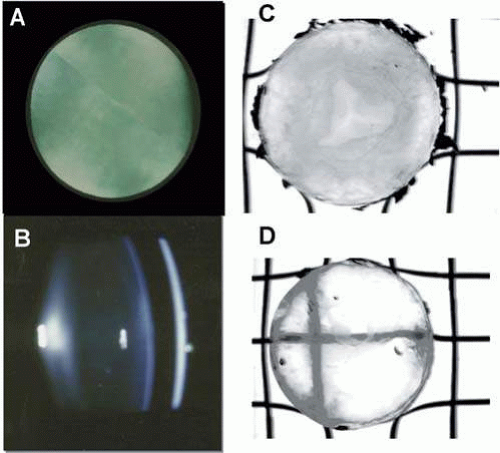 Figure 18.8. Aldose reductase in the dog lens. Antibodies for AR, visualized with DAB-PAP, are present in the epithelium in A, and superficial cortical fibers at the bow region, B. C. The early vacuolar stage of cataract by slit lamp (left) and magnetic transfer contrast magnetic imaging (MTC-MRI, right). D. A superficial cortical opacity with water clefts. E. A full cortical cataract. Using MTC-MRI, lighter shades represent increased tissue/protein hydration. Note the appearance of water vacuoles (C, arrows), hydration of the anterior superficial cortex (D, arrow), increasing liquefaction in the bow region (E, arrows), and increasing hydration of outer to inner cortex (D, E). |
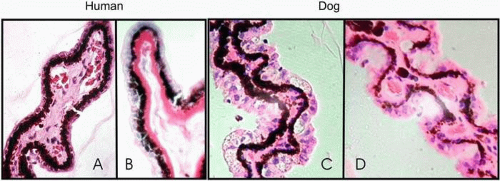 Figure 18.9. Retroillumination (A) and slit lamp (B) photograph of the lens from a 38-month galactose-fed beagle orally treated with the ARI, M79175, from the onset of galactose feeding. The photographs demonstrate the absence of lens opacities in this prevention study. E and F represent lenses obtained from an intervention study in which all dogs received a galactose diet for 6 months. After 3.5 months, when all dogs developed cortical cataracts, each dog was topically administered either the ARI, Kinostat, or vehicle for the remainder of the study. C. Lens from a vehicle-treated dog. D. The appearance of a Kinostat-treated dog lens obtained at the end of the study. Note the appearance of the dense mature cataract in C compared with the treatment improved lens in D, indicating that the cortical cataract had been partially reversed. |
Biochemical studies indicate that in vitro incubated dog lenses rapidly reduce 3-fluoro-3-deoxyglucose and 3-fluoro-3-deoxy-galactose to 3-fluoro-3-deoxysorbitol and 3-fluoro-3-deoxygalactitol, respectively. Moreover, this reduction is inhibited by the addition of ARI.211 Sugar alcohol formation in lens epithelial cells induces apoptosis, which also is prevented by the addition of an ARIs.102 A similar increased apoptosis of lens epithelial cells has been reported in lenses extract from diabetic patients.212 The osmotic nature of canine sugar cataracts has been confirmed by both histology208 and through magnetization transfer contrast magnetic resonance imaging (MTC-MRI), a technique that can determine the water environment around proteins.213 MRI-MTC demonstrated that distinct changes in the size and shape of the lenses occur during sugar cataract formation.214 Therefore, although no direct evidence for the osmotic hypothesis in human diabetic cataracts exists, the number of similarities between human and animal lenses suggest that AR is involved. The production of sorbitol confined to localized areas containing increased levels of AR, as suggested by the localization and Raman spectroscopy data, could conceivably exert sufficient osmotic stress to initiate cataract formation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


