Ocular Manifestations of the Rheumatic Diseases
Anat Galor
Jennifer E. Thorne
Douglas A. Jabs
The rheumatic diseases are a heterogeneous collection of diseases that are multisystem in nature and immunologically mediated. They are loosely grouped into three general categories: the arthritides, the connective tissue diseases, and the vasculitides (Table 26-1). Behçet disease has been classified variously but in this chapter is discussed with the vasculitides. The diagnosis of each of the rheumatic disorders is a clinical diagnosis. To ensure homogeneity in clinical research and assist in reporting in the literature, the American College of Rheumatology (ACR) has developed classification criteria for most of the rheumatic diseases. These classification criteria are for research purposes and cannot always be used in the care of every patient. However, they often provide a useful description of the clinical features of the disease.
Table 26-1. The Rheumatic Diseases | |
|---|---|
|
The clinical features of the rheumatic disorders are based on the anatomic pattern of disease. This situation is analogous to the definitions of ocular uveitis syndromes, in which the anatomic pattern of disease is used as characteristic criteria. Thus, rheumatoid arthritis generally is described as an additive, symmetric, deforming polyarthritis, whereas the seronegative spondyloarthropathies often are associated with a migratory, asymmetric oligoarthritis. Similarly, the different vasculitides are defined by the pattern of vascular involvement and the size of blood vessels commonly involved. As such, individual laboratory tests (e.g., rheumatoid factor [RF] or antinuclear antibody [ANA]) are supporting evidence in the diagnosis of the disease but do not define the disease.
The major ophthalmic manifestations of the rheumatic diseases include scleritis, Sjögren syndrome, uveitis, retinal vascular disease, and neuroophthalmic lesions.1,2 Each of these is most characteristically associated with a few but not all of the rheumatic disorders. Hence, scleritis most often is seen with rheumatoid arthritis or with vasculitis. Acute anterior uveitis most often is seen with the seronegative spondyloarthropathies. Retinal vascular and neuroophthalmic lesions are seen with disorders that have either a vaso-occlusive component, such as systemic lupus erythematosus or one of the vasculitides.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is the most common rheumatic disorder, affecting an estimated 1% of the general population.3,4,5,6,7 The overall incidence of RA has remained fairly stable across geographic locations.3 Classically RA is described as an additive, symmetric, deforming polyarthritis. The diagnosis of RA is a clinical one, based on the features outlined in the paragraphs that follow. The ACR classification criteria for RA are outlined in Table 26-2.8
Table 26-2. American College of Rheumatology (ACR) Revised Criteria for the Diagnosis of Rheumatoid Arthritis | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Etiology and Pathogenesis
The etiology of RA remains unknown, although both genetic and environmental factors likely are involved.9 Theories on the pathogenesis of RA postulate an inciting immunologic event in a genetically susceptible individual with subsequent inflammation in the joint resulting in synovial proliferation and joint destruction. The initial inciting immunologic event is unknown, but both immune complex disease and cellular immune events have been hypothesized.10,11 Immunohistologic studies of the joint space have shown an early accumulation of T lymphocytes, which localize to the synovial tissue and initiate the inflammation that causes the clinical manifestations of RA.12 Proliferation of synovial fibroblasts, synovial hypertrophy, and pannus formation occur.9,12 In established RA there is an accumulation of polymorphonuclear leucocytes (PMNs) in the synovial fluid, suggesting recruitment of PMNs in later stages of the disease. These changes lead to erosion of bone at the joint margin, osteopenia at the joint margin, and in long-standing disease, joint destruction.
These immunologic processes cause the release of cytokines and metalloproteinases that perpetuates the joint destruction and leads to the production of serum RF.9,12 RF is a polyclonal autoantibody directed against immunoglobulin G (IgG), which results in circulating immune complexes. It can be detected by several tests, including sheep red blood cell agglutination and enzyme-linked immunosorbent assay (ELISA). RF is a useful marker for the presence of RA and is present early in the disease course in 85% to 90% of patients with RA.6,12 However, its contribution to the pathogenesis of the disease remains unclear, and the presence of RF is neither necessary nor sufficient for the diagnosis of RA.6,12 High levels of RF have been associated with more severe clinical disease and poorer prognosis.12,13 Anticitrullinated peptide (ACP) antibodies have also been reported in the serum of patients with RA.9 ACP antibody positivity has a high specificity (∼ 90%) for RA, has a sensitivity similar to RF, and levels can be detected before the start of clinical disease. Similar to RF, although these antibodies predict a more aggressive disease course, their participation in disease pathogenesis is unclear.9
There is a genetic predisposition to RA, with an association of the disease with the human leukocyte antigen (HLA) types, HLA-DR4 and HLA-DR1.14 Both HLA-DR4 and HLA-DR1 are thought to be involved in both the susceptibility to and the severity of RA.12 In population studies, more than 90% of patients with RA have HLA-DR4 or HLA-DR1 or both.14 Polymerase chain reaction techniques have identified a specific sequence in the DRβ 1 gene that encodes an amino acid motif or shared epitope that has been found to occur in both HLA-DR4 and HLA-DR1 haplotypes.12 This shared epitope has been hypothesized to serve as either a binding site for an antigenic peptide or as an autoantigen that activates T-cell inflammation.12 Other genetic polymorphisms have also been implicated in RA susceptibility including polymorphisms in the genes for tumor necrosis factor α (TNF-α), PTPN22, and peptidyl arginase deiminases.9
Clinical Features
In most patients with RA, the onset is insidious. However, in 10% to 20% of patients there may be an abrupt or explosive onset. The arthritis classically is described as an additive, symmetric, deforming polyarthritis. Typically, the small joints of the hands and feet are involved, although all joints may be affected. As with all inflammatory arthropathies, the disease is characterized by a gel phenomenon, that is, stiffness at rest improving with movement. Most patients complain of morning stiffness. X-ray studies of the affected joints show erosions of bone at the joint margin, which evolve into joint destruction with long-standing disease. Because of the damage to surrounding structures, joint laxity may develop; this process results in the characteristic ulnar deviation of the fingers.6
Extra-articular features are common in patients with RA.6,8,9,10,11,12,13,14,15,16 Generally, the number and severity of extra-articular complications increases with increasing disease duration and severity.6 The most common extra-articular lesion is the presence of rheumatoid nodules. These nodules occur in approximately 25% of patients with RA and are located primarily on extensor surfaces. On biopsy, they have a characteristic histologic picture with central fibrinoid necrosis surrounded by a palisade of elongated histiocyte-like cells. This core is enveloped by an outer zone of mononuclear inflammatory cells, primarily lymphocytes and plasma cells. Immunohistochemical analysis has demonstrated that the cells within the palisade layer stain for monocyte markers and that peripheral lymphocytes are predominantly T cells.17
Pulmonary lesions that may occur include pleural disease, pulmonary effusions, pleural or pulmonary nodules, arteritis with resultant pulmonary hypertension, and interstitial fibrosis. Pleuritis develops in 20% of patients with RA and may occur concurrently with the initial arthritic symptoms. Pulmonary effusions may be large enough to cause dyspnea. Rheumatoid nodules may develop in the pleura or within the lung. In the lung, the nodules may develop in clusters and cavitate, leading to bronchopleural fistula.6
Cardiac involvement includes pericarditis, which may be detected in as many as 50% of patients with RA at autopsy. Pericardial effusion secondary to pericarditis may be seen on echocardiogram in up to 31% of patients with RA, although most patients are asymptomatic and left ventricular dysfunction because of the effusions is rare.6,18 Cardiac conduction defects as a consequence of rheumatoid nodules present in the conducting system can occur. Rheumatoid nodules also have been detected on heart valves.6,16
Rheumatoid vasculitis occurs in less than 1% of patients with RA and typically is seen in patients with severe arthritis and high RF titers. It presents as a polyneuropathy, particularly with lower extremity anesthesia or hypesthesia, poorly healing leg ulcers, or as palpable purpura. Digital gangrene and occasionally visceral ischemia also may be manifestations of rheumatoid vasculitis.6,16,19,20
Felty syndrome consists of RA, splenomegaly, and leukopenia and may represent up to 3% of patients with RA.21 Rheumatoid factor is present in 98% of patients with Felty syndrome, and titers typically are high. The pathogenesis of the leukopenia is unknown, but possible mechanisms include accelerated removal of leukocytes and suppression of granulopoiesis. Chronic leg ulcers with recurrent infections and hyperpigmentation of the skin also are characteristic features.6,22
Treatment
The goals of treatment for RA include the prevention and control of joint damage, prevention of loss of function, and pain reduction.23 Prior to the 1990s, the treatment of RA typically was approached in a stepwise fashion with nonsteroidal anti-inflammatory drugs (NSAIDs) being the first line of therapy followed by institution of disease-modifying antirheumatic drugs (DMARDs) in more severe or refractory cases.23 However, recent studies suggest that the destruction of the joints and loss of function occurs soon after the onset of synovitis.24,25 It is estimated that one half of the joint destruction that is seen over an 8-year period occurs in the first 2 years.26 Therefore, now a DMARD typically is instituted on diagnosis of RA in addition to an NSAID.24 This new treatment paradigm has resulted in a significantly improved prognosis for patients with RA with increased joint preservation and improved functioning.23 NSAID therapy includes salicylates and cyclooxygenase (COX) inhibitors. These NSAIDs are used to control pain and swelling and to improve the joint function but typically do not alter disease course or prevent joint destruction long term.23 Because joint destruction can occur within the first 2 years of the disease, early treatment with a DMARD or remittive agent is recommended.24 Remittive agents include methotrexate (MTX), sulfasalazine (SSZ), leflunomide, and various biologic therapies (etanercept, infliximab, adalimumab, abatacept, rituximab). Methotrexate, an antimetabolite, typically is the first choice of remittive agents and has been found to be efficacious in the treatment of RA in multiple randomized clinical trials.27,28,29 Methotrexate was found to improve clinical signs and symptoms of RA, improve functional status, and slow radiographic disease progression. Leflunomide also has been found to be effective in controlling RA progression and in improving clinical and functional status both as monotherapy and in combination with methotrexate.30,31,32 Combination therapy with various agents (e.g., MTX, hydroxychloroquine, and SSZ) has also been shown to be effective but is less commonly used due to toxicity.33,34
Biologics are engineered agents designed to block cytokines or cytokine receptors. Biologic agents, including anti–tumor necrosis factor α α (TNF-α α) agents (etanercept, infliximab, adalimumab, golimumab, certolizumab), abatacept, and rituximab have been found to be potent remittive agents in several randomized clinical trials, mostly when used in conjunction with MTX.23,24,35,36,37,38,39 Etanercept is a recombinant TNF-α α receptor Fc segment fusion protein and is effective in controlling progression of RA as initial monotherapy in early RA and as therapy for patients who have failed previous DMARD therapy.23,24,35,36 The typical dose of etanercept is 50 mg administered subcutaneously weekly. Many patients taking etanercept improve more rapidly than those taking methotrexate, sometimes within 2 weeks after instituting etanercept.24,36 Etanercept also is effective in combination with methotrexate for controlling ongoing active RA in patients already taking methotrexate alone.36 Infliximab is a chimeric (mouse-human) anti–TNF-α α monoclonal antibody and is available only as an intravenous preparation given 3 to 10 mg every 4 to 8 weeks. Infliximab in combination with methotrexate has been used successfully to improve clinical and functional status in patients with RA who have failed methotrexate monotherapy.37,38 Adalimumab is a fully humanized anti-TNF-α α monoclonal antibody and is available as a subcutaneous injection given biweekly at a dose of 40 mg. Adalimumab, in combination with methotrexate, has been shown to improve clinical signs and symptoms and slow radiographic disease progression of RA.23,40 The anti-TNF-α α agents are the most commonly used biologic medications in the treatment of RA; newer biologic agents are mostly reserved for cases where there is an inadequate response to these first-line therapies.
Abatacept, a fusion protein composed of an immunoglobulin fused to the extracellular domain of CTLA-4, inhibits the costimulation of T cells.23 It is given as a monthly intravenous infusion of 10 mg/kg. Rituximab, a chimeric monoclonal antibody against CD20 (found on peripheral B cells), is also given as an intravenous infusion (1,000 mg on days 1 and 15). Both abatacept and rituximab, in combination with methotrexate, have been shown to improve clinical signs and symptoms, slow radiographic disease progression, and improve function in patients with RA.23,41,42
Anakinra, a recombinant human interleukin-1 receptor antagonist (IL-1Ra), also is approved for use in RA in a 100-mg daily dose given by subcutaneous injection.24 Anakinra inhibits the binding of the cytokines IL-1α and IL-1β to the IL-1 receptor, which prevents the activation of its target cells and subsequent synovial inflammation and joint destruction.24 Randomized clinical trials found anakinra to be superior to placebo in controlling clinical symptoms of RA.24 Another randomized clinical trial found that anakinra in combination with methotrexate was more effective in controlling RA progression than methotrexate alone.39
Immunosuppressive drugs, such as azathioprine43,44 or cyclosporine,45 are used less frequently because of toxicity. Cyclophosphamide may be used in the treatment of rheumatoid vasculitis.44,46 Traditionally, low-dose prednisone (5 mg every other day to 10 mg/d) was used to decrease stiffness and increase mobility. Although the newer remittive agents have decreased this practice, prednisone still is used for symptomatic relief.
Ocular Manifestations
The most common ocular manifestations of RA are keratoconjunctivitis sicca (secondary Sjögren syndrome), scleritis, and rheumatoid corneal melts.2,47 Sjögren syndrome originally was defined as dry eyes, dry mouth, and RA. It subsequently has become evident that Sjögren syndrome can exist in association with a connective tissue disease, in which case it is termed secondary Sjögren syndrome, or by itself with no definable associated connective tissue, in which case it is termed primary Sjögren syndrome. In Sjögren syndrome, there is a lymphocytic infiltration of the lacrimal and salivary glands resulting in glandular destruction and dysfunction. This process ultimately results in a characteristic loss of tearing and saliva. Approximately 11% to 13% of patients with RA have secondary Sjögren syndrome.2
Scleritis (Fig. 26-1) is the second most common ocular manifestation of RA, affecting an estimated 1% to 6% of patients with RA and 14% of patients with rheumatoid vasculitis.2,48,49 Scleral inflammation can be classified as episcleritis or scleritis. Episcleritis presents with discomfort rather than pain, more superficial ocular inflammation, less frequent and severe ocular complications, and typically a less frequent association with systemic disease.50 Conversely, scleritis often is characterized by pain, presents with deeper inflammation and edema in the sclera, often has ocular complications, which may be severe, and in approximately one half of the cases, is associated with systemic disease.50,51 Episcleritis may be self-limited and spontaneously remitting, whereas scleritis typically requires therapy. Scleritis may be classified as anterior, posterior, or necrotizing. Anterior scleritis is subdivided into diffuse and nodular types. Scleromalacia perforans is a separate category, in which there is an insidious but destructive scleral process; it is seen in patients with long-standing RA. Any of the previously named types of scleritis may be seen in association with RA, although anterior scleritis is most common.2,50
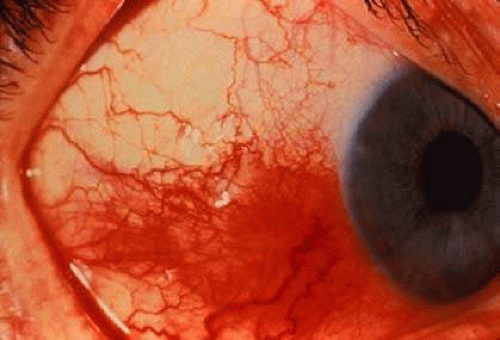 Fig. 26-1. Scleritis in a patient with rheumatoid arthritis. (Courtesy of David Knox, MD.) |
Although estimates of the frequency of scleritis in patients with RA have been as high as 6%, large series have shown that approximately 1% of patients with RA have scleral disease.2,49,52 Of patients with scleritis, 10% to 33% have RA, and RA represents the most commonly seen rheumatic disease in patients with scleritis.47,50 In patients with RA, scleritis is associated with more severe systemic disease and with a greater frequency of extra-articular manifestations.47 Patients with RA are more likely to develop necrotizing scleritis than those patients with idiopathic scleritis.53 Necrotizing scleritis is more likely to be associated with systemic vasculitis and a poor systemic prognosis. In a retrospective study by Foster et al,48 29% of patients with RA and necrotizing scleritis or necrotizing keratitis died, typically because of complications of vascular disease.
Rheumatoid corneal melts, also known as marginal corneal ulcers, may be either bland (not inflammatory) or necrotizing (peripheral ulcerative keratitis [PUK]) in nature. These melts have been described as occurring spontaneously or after ocular surgical procedures.54 Noninflammatory marginal keratitis often can be treated with local measures. Necrotizing keratitis requires aggressive medical therapy with systemic corticosteroids and often immunosuppressive drugs. Patients with PUK and RA have a high mortality rate, possibly because of an underlying rheumatoid vasculitis.2,48,49,53
Other uncommon features include Brown syndrome,55,56 orbital myositis,57 and hydroxychloroquine retinal toxicity.58,59 Brown syndrome has been described in a few patients with RA and is caused by a stenosing tenosynovitis of the superior oblique tendon.55,56 Rare cases of a retinal microangiopathy with cotton-wool spots have been reported,2 but these probably are the result of an associated rheumatoid vasculitis.
Seronegative Spondyloarthropathies
The seronegative spondyloarthropathies include ankylosing spondylitis (AS), Reiter syndrome, arthritis with inflammatory bowel disease, and psoriatic arthritis. Historically, these disorders were called seronegative because they lacked rheumatoid factor, which distinguished them from RA, in which patients were said to be seropositive. The seronegative spondyloarthropathies are better termed the HLA-B27–associated spondyloarthropathies, because of their statistical association with the gene HLA-B27. In addition, these disorders are linked by several clinical features and sometimes are difficult to differentiate in the earlier stages of the disease. Clinical findings include a predilection for axial skeletal involvement, extra-articular features, enthesopathy (inflammation of the insertion of tendons), and an asymmetric, migratory oligoarthritis.60 The most common ocular manifestation of the seronegative spondyloarthropathies is a nongranulomatous, recurrent, acute anterior uveitis. Acute anterior uveitis shares with the spondyloarthropathies an association with the gene HLA-B27; approximately 50% of patients with acute anterior uveitis possess HLA-B27,61,62,63 and if the uveitis is unilateral and recurrent, up to 71% of patients will be HLA-B27 positive.63 Approximately 50% of patients with HLA-B27–associated acute anterior uveitis have a seronegative spondyloarthropathy.63,64
Etiology and Pathogenesis
The seronegative spondyloarthropathies (Table 26-3) are thought to be multifactorial in nature and triggered by an environmental insult in a genetically predisposed person. This genetic predisposition is evidenced by the strong association with HLA-B27.61,65,66,67,68,69 AS has the strongest association in that more than 90% of white patients with AS are HLA-B27 positive, whereas the general population frequency of HLA-B27 is approximately 8%.2,61,65
Table 26-3. The Seronegative (HLA-B27–Associated) Spondyloarthropathies | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
The best insight into a possible environmental trigger for these disorders is provided by epidemic reactive arthritis. Epidemic reactive arthritis occurs after an infectious gastroenteritis with a limited number of organisms, including Shigella, Salmonella, and Yersinia. The subsequent arthritis occurs well after the infectious gastroenteritis has resolved and is a sterile arthritis. The arthritis appears to represent an immunologic response to the infectious organism, which triggers the arthritis in a susceptible person. In reactive arthritis, the presence of HLA-B27 again is associated strongly with the development of disease.60,70
The exact molecular mechanisms by which HLA-B27 predisposes to disease are unknown. Theories include molecular mimicry60,71 and the possibility that HLA-B27 is associated with a defective class I antigen-mediated cellular response.69 The molecular mimicry hypothesis suggests that there is a similarity at the molecular level between the HLA-B27 molecule and the inciting organisms, allowing for the triggering of an immune response and the subsequent development of clinical disease.69,71 The process is felt to be analogous to rheumatic fever or acute poststreptococcal glomerulonephritis, where the immune response to a cross-reactive antigen results in a subsequent immune-mediated disease. Alternatively it has been suggested that the HLA-B27 molecule may be a defective molecule associated with an aberrant cytotoxic T-cell response.69,72
Ankylosing Spondylitis
Clinical Features
AS is characterized by involvement of the axial skeleton and bony fusion (ankylosis).73 The estimated prevalence of AS is 0.1% to 0.2% of the general population and is 2% in HLA-B27–positive patients.69 Young adult males are affected most frequently, with a male-to-female ratio of approximately 3:1. Earlier studies suggested a much higher male-to-female ratio, but it subsequently has become evident that women may have mild or atypical forms of the disease.74,75 Between 10% and 30% of HLA-B27–positive first-degree relatives of AS patients will develop the disease.76
The characteristic symptom of AS is chronic low back pain, which improves with exercise. The symptoms may be mistaken by patients as a strain-related injury, and the diagnosis may be delayed by several years.69,77 As with other inflammatory arthropathies, the disease is associated with morning stiffness. The inflammation results in fusion of the axial skeleton (spinal ankylosis) and sacroiliitis. Physical examination reveals restricted motion of the spine and may reveal tenderness over the sacroiliac joints. Radiographic examination of the spine and sacroiliac joints is important in the diagnosis of the disease. The demonstration of sacroiliitis on radiographic examination of the sacroiliac joints is almost a sine qua non for the diagnosis of spondylitis. The end stage of this process is a completely fused and immobilized spine, also known as a bamboo or poker spine. In addition to the spinal arthritis, patients may also develop an arthritis of the shoulders and hips, limited chest expansion, restrictive lung disease, apical pulmonary fibrosis, aortic insufficiency because of arteritis, and heart block.60,73,78
Treatment
For most patients, AS is a chronic, slowly progressive disease. As such, treatments focus on relieving pain and stiffness and maintaining good posture. NSAIDs and a program of physical therapy and exercise are used to achieve these goals.73 Although NSAIDS do not reverse and generally do not prevent the ultimate spinal ankylosis, they do relieve symptoms and allow patients to participate in an exercise program. A program of physical therapy and exercise is important as it allows patients to maximize the mobility of the spine and to allow spinal fusion to occur in a posture most advantageous to the patient.60 Immunosuppressive agents (including MTX and SSZ) have not been shown to affect the course of axial disease.73 Phase III clinical trials have demonstrated that etanercept, infliximab, and adalimumab are beneficial in AS in patients with persistently high disease activity (both axial and peripheral).73 As such, current treatment paradigms typically use an anti-TNF agent in such patients, most often in combination with a MTX in peripheral disease or as monotherapy in axial disease. 73
Ocular Manifestations
The primary ocular manifestation of AS is a nongranulomatous, recurrent, acute, unilateral or unilateral alternating anterior uveitis.64,76 Generally, one eye is affected at a time, although both eyes may suffer attacks.64,76 Occasionally anterior chamber inflammation may be severe enough to produce hypopyon, fibrin deposition, and posterior synechiae (Fig. 26-2).78 An estimated 25% to 40% of patients with AS suffer an attack of uveitis at some time during the course of their disease, with eye inflammation occasionally preceding the systemic diagnosis.69,76,79 Studies of patients with acute anterior uveitis have reported that AS is present as the underlying systemic disease in 18% to 34% of these patients.76,78,80,81 Rarely, a spillover vitritis is seen in patients with acute anterior uveitis and AS, but it is substantially less frequent than the typical iridocyclitis.82 Scleritis has rarely been reported in patients with HLA-B27–associated disease.83
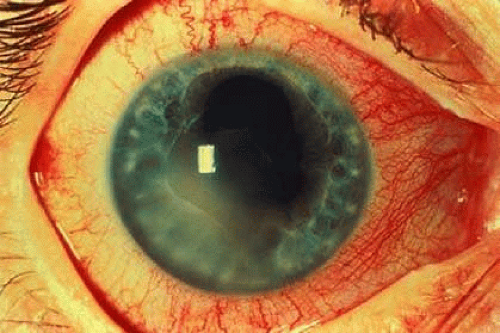 Fig. 26-2. Fibrin clot and posterior synechiae in a patient with acute, anterior uveitis and ankylosing spondylitis. |
Undifferentiated Spondyloarthritis and Reactive Arthritis
Clinical Features
Reiter syndrome was originally characterized by the classic triad of arthritis, urethritis, and conjunctivitis. Typically, it is rare for a patient to present with the classic triad originally described. As such, the term Reiter syndrome has been replaced with the entities undifferentiated spondyloarthritis and reactive arthritis. Undifferentiated spondyloarthritis refers to patients with inflammatory spinal pain and/or synovitis but without evidence of AS.84 Enthesitis (i.e., inflammation at the site of an insertion of a ligament, tendon, or fascia to bone) is another common finding. The clinical course of undifferentiated spondyloarthritis varies per individual, with some experiencing disease remission, some progressing to AS, and some continuing with undifferentiated inflammation. As in AS, first-line treatment is with NSAIDs, with methotrexate and TNF inhibitors reserved for more severe cases.84
Reactive arthritis refers to patients who develop an inflammatory arthritis after an infection, with symptoms typically starting 1 to 4 weeks after an episode of gastroenteritis or urethritis.84 The microbials associated with reactive arthritis include the enteric bacteria Salmonella, Shigella, Yersinia, Campylobacter, Clostridium difficile, and Chlamydia (whose various species can cause urethritis and upper respiratory infections).84 The arthritis of reactive arthritis is migratory, asymmetric, episodic oligoarthritis affecting primarily the large joints at the lower extremities, such as the knees or ankles. Other articular features include heel pain, sausage digits caused by interphalangeal arthritis of the toes and/or fingers, and sacroiliitis. Mucocutaneous lesions include urethritis in men and cervicitis in women, circinate balanitis, painless oral ulcers, keratoderma blennorrhagica, and dystrophic nail lesions.84,85 Systemic symptoms including fever and weight loss also may occur. The disease tends to follow an episodic and relapsing course.84,85,86 The arthritis may be recurrent in 15% to 50% of patients.84 Another 15% to 30% of patients will develop chronic arthritis over 10 to 20 years of follow-up.84,86
Ocular Manifestations
Conjunctivitis was part of the original triad described by Reiter and is one of the hallmarks of the disease. It tends to be a feature of early disease, particularly of the initial attack, and may be missed if patients are seen only during subsequent attacks. Depending on the source of the reported series, and on the diagnostic criteria used, the frequency of conjunctivitis in patients with Reiter syndrome has varied from 33% to 100%. The more serious ocular manifestation is recurrent, acute, nongranulomatous anterior uveitis. It occurs in approximately 5% to 20% of patients with the initial attack but may occur in as many as 50% of patients with reactive arthritis over long-term follow-up.69,76,87 Some studies of patients with acute anterior uveitis suggest that reactive arthritis is more frequently seen in patients with acute anterior uveitis than is AS,63,88 although this finding may be the result of a referral bias or of geographic variations in the prevalence of these disorders. As with AS, occasionally a spillover vitritis has been described in reactive arthritis and is found more commonly with reactive arthritis than with AS. Rarely other forms of posterior ocular involvement such as multifocal choroiditis and panuveitis have been reported.89,90 Optic disc edema in the setting of an anterior uveitis has also been described.2 Rarely, patients may develop a keratitis with punctate epithelial lesions, progressing to a central loss of the corneal epithelium and subepithelial infiltrates.91,92,93
Arthritis with Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) consists of two distinct diseases: ulcerative colitis (UC) and Crohn disease. Ulcerative colitis is an inflammatory disorder of the colonic mucosa. Crohn disease, also known as regional enteritis, granulomatous ileocolitis, or granulomatous colitis, is a focal granulomatous disease involving any area of the gastrointestinal tract.
Clinical Features
The clinical features of IBD are diarrhea and abdominal pain. Diarrhea may or may not be bloody. Extraintestinal manifestations of IBD include dermatitis, mucous membrane disease, ocular inflammation, and arthritis. Skin disorders occur in 6.5% to 15% of patients with IBD and include erythema nodosum and pyoderma gangrenosum.94,95 Arthritis occurs in up to 25% of patients with IBD.96 One form of arthritis associated with IBD is an HLA-B27–associated axial-sacroiliac spondylitis, which is seen in approximately 20% of patients with Crohn disease and in 10% to 15% of patients with ulcerative colitis.96 In one series of 103 patients with IBD, arthritis occurred in 39% of patients, and of the patients with arthritis, 90% met the criteria for spondyloarthropathy.97 Between 50% and 70% of patients with spondylitis and IBD are HLA-B27 positive.96 Of the HLA-B27–positive patients with IBD, 50% will develop acute anterior uveitis.94 The activity of this spondylitis is unrelated to that of the bowel disease, and in 18% to 20% of patients, the spondylitis will be asymptomatic.69,96,98 Another form of arthritis seen in patients with IBD involves the peripheral joints and is seen in 5% to 15% of patients.96 Type 1 disease is a large-joint, lower-extremity, nondeforming oligoarthritis that tends to have an acute onset and resolves within 6 weeks. Type 2 disease is an arthritis involving more than five joints and tends to have a more chronic course. There tends to be a correlation between peripheral arthritis and gut activity in UC but not in Crohn disease.96
Treatment
Treatment of the IBD is individualized and dependent on the extent and severity of the disease. Typically, treating the underlying bowel disease improves the joint disease. Localized proctitis often may be treated with corticosteroid enemas. More extensive bowel disease may require systemic corticosteroid therapy. Generally, nonsteroidal therapy with sulfasalazine is used first in an effort to minimize steroid complications and often is used to decrease the dose of corticosteroids needed. Severe corticosteroid-dependent IBD also can be treated with corticosteroid-sparing agents, such as azathioprine or methotrexate.96,98 Infliximab has been shown to induce remissions in Crohn disease and is effective in the treatment of Crohn-related fistulas.99 However, less evidence exists for the benefit of infliximab in ulcerative colitis.96 Because of the risk of colonic malignancy with long-standing ulcerative colitis, a colectomy is sometimes performed. After colectomy the mucocutaneous manifestations and the enteropathic arthritis may resolve in some patients (∼ 50%), but paradoxically, the arthritis has also been reported to begin after surgery.96
Ocular Manifestations
Ocular inflammation has been reported to occur in up to 13% of patients with IBD but typically occurs in approximately 2% to 6% of patients.76,84,98 The ocular manifestations include anterior uveitis, scleritis, and keratitis. Anterior uveitis is the most common ocular manifestation.100,101,102 More often it presents as a nongranulomatous, recurrent, acute anterior uveitis. This type of uveitis is seen in association with spondylitis and HLA-B27. Chronic and bilateral uveitis also occurs occasionally and may be seen more frequently in women with IBD.76,103,104 There have been occasional case reports of retinal vascular disease, including retinal artery occlusion and retinal vasculitis, and ischemic optic neuropathy in patients with IBD.105,106,107 It has been suggested that these retinal vascular lesions are caused by a thrombic diathesis seen in some patients with IBD.105 All types of scleritis have been associated with IBD including anterior scleritis, necrotizing scleritis, and posterior scleritis. The scleral inflammation may parallel the activity of the underlying bowel disease (Fig. 26-3).101,102 Rarely, keratitis102,108 and Brown syndrome109 may be seen in association with IBD.
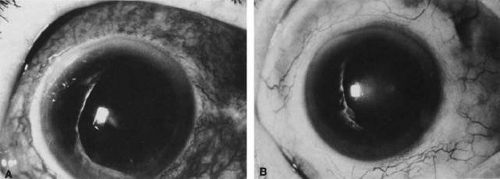 Fig. 26-3. Scleritis in a patient with inflammatory bowel disease. A. Active scleritis. B. Healed scleritis after treatment of the inflammatory bowel disease. |
Psoriatic Arthritis
Clinical Features
Psoriatic arthritis is a syndrome defined by the presence of psoriasis and an associated seronegative inflammatory arthritis.110 Psoriasis is a common cutaneous disorder, affecting 1% to 3% of the population and is characterized by erythematosus, well-demarcated macular lesions with silvery scales. It characteristically occurs on extensor surfaces, particularly the elbows and scalp, but also the chest or back. Approximately 10% to 20% of patients with psoriasis have arthritis.110,111 The skin disease generally precedes the onset of the arthritis by several years, but the arthritis precedes the onset of skin disease in 15% of patients with psoriatic arthritis.110 Nail changes are present in 90% of patients with psoriatic arthritis as compared to 41% of patients with psoriasis but without arthritis.112 The nail changes include pitting, transverse ridges, crumbling, and onycholysis.
Psoriatic arthritis has been classified into five groups based on the clinical presentation of the arthritis. Classic psoriatic arthritis is manifested by involvement of the distal interphalangeal joints. Arthritis mutilans is a severely deforming, destructive, and usually widespread arthritis with ankylosis of joints and characteristic erosive joint changes seen radiographically. A symmetric, additive, deforming polyarthritis similar to RA can be seen. However, psoriatic arthritis usually is negative for RF. The most common presentations of psoriatic arthritis are monoarthritis or an asymmetric oligoarthritis, usually affecting the distal interphalangeal, proximal interphalangeal, or metatarsal interphalangeal joints. The fifth type of psoriatic arthritis is psoriatic spondylitis. Psoriatic spondylitis is an HLA-B27–associated disorder, and approximately 50% of patients with psoriatic spondylitis are HLA-B27 positive.60,110 The course and severity of disease in psoriatic arthritis appears to be independent from the skin disease.110
Ocular Manifestations
Conjunctivitis occurs in approximately 20% to 33% of patients with psoriatic arthritis, iritis in 4% to 7%, and scleral disease in 2%.60,110,111,113 Patients with psoriatic spondylitis typically develop a nongranulomatous, recurrent, acute anterior uveitis, as is the case with other HLA-B27–associated arthropathies, but chronic uveitis also may occur. Brown syndrome also has been reported in patients with psoriatic arthropathy.114
Juvenile Idiopathic Arthritis
Juvenile idiopathic arthritis (JIA), also known as juvenile chronic arthritis (JCA) or juvenile rheumatoid arthritis (JRA), is defined as an arthritis of one or more joints lasting at least 6 weeks in patients 16 years of age or younger.115 Other causes of arthritis in children, such as leukemia, must be excluded. The traditional classification of JIA depends on the pattern of presentation of the arthritis and has been classified as polyarticular, oligoarticular, and systemic disease. Polyarticular JIA is defined as presentation with five or more joints involved and is seen in 30% of children presenting with JIA. Oligoarticular JIA involves four or fewer joints and comprises 60% of children with JIA. Systemic disease is the disorder originally described by Still, affects 10% of children with JIA, and has prominent systemic features with a variable arthritis.116 More recent classifications have been proposed;he most often used being the International League of Associations for Rheumatology (ILAR) Classification.116
Clinical Features
The incidence and prevalence of JIA have been estimated as 10 to 20 cases per 100,000 population per year and 65 to 148 cases per 100,000 population, respectively.117,118 Five distinct subgroups of JIA have been described. The systemic variant of JIA also is known as Still disease. The male-to-female ratio is approximately 1:1, and the onset of disease is generally at younger than 5 years of age. The arthritis has a variable relation to the onset of the systemic disease but is usually a polyarthritis. The systemic features include fever, a salmon-colored evanescent maculopapular rash, lymphadenopathy, hepatitis, splenomegaly, and serositis. An elevated erythrocyte sedimentation rate and leukocytosis are seen, but ANA and RF tests are negative in at least 90% of cases.116 Ocular disease generally is not associated with this variant.115
Two types of oligoarticular JIA have been described. Persistent oligoarthritis affects four or fewer joints through the disease course whereas extended oligoarthritis affects five or more joints after the first 6 months of the disease.116 Oligoarticular disease is typically seen in children younger than 5 years of age. The mean age of onset is 3 years. Girls are affected more often than boys with a male-to-female ratio of 1:4. The arthritis is an oligoarticular, large-joint, lower-extremity arthritis that tends to spontaneously remit and not leave significant articular deformities. Approximately 60% of these patients have a positive ANA test. Several loci of genetic susceptibility have been found in this subgroup of patients including ones involving the HLA alleles at class I (HLA A2) and class II (DRB1*0801, DRB1*1104. DRB1*1301).116,119,120 A chronic anterior uveitis is the characteristic ocular feature of this group seen in 20% to 30% of children.115,121,122,123 A second type of oligoarticular disease (classified as enthesitis-related arthritis) is seen in older children, with a mean age of onset at 12 years. Boys are affected more often than girls, and the male-to-female ratio is approximately 7:1.116 The arthritis is a large-joint, lower-extremity oligoarthritis. Sacroiliitis is common, and these children tend to progress to AS over time. Enthesitis is also a common finding. Eighty percent of these children are HLA-B27 positive, and approximately 25% develop a nongranulomatous, recurrent, acute anterior uveitis. This variant shares the clinical and immunogenetic features of the seronegative spondyloarthropathies in adults, including the same type of uveitis.115,124,125
Two types of polyarticular disease have been described. In children older than 10 years of age, an RF-positive (seropositive), rheumatoid-like polyarthritis may develop. Girls are affected more commonly than boys with a ratio of 9:1. The arthritis is an additive, symmetric, deforming polyarthritis similar to that of adult RA. The arthritis is persistent and results in long-term articular morbidity with joint erosions and deformity seen. This type of arthritis is strongly associated with HLA-DRB1*0401, the HLA antigen seen in adult RA. Ocular disease is uncommon in this subgroup.115,116,124 A second type of polyarticular disease is one in which the RF factor is negative (the ANA is positive in approximately 40% of patients).116 The peak age of disease onset is between 6 and 7 years; the arthritis is variable and may be symmetric or asymmetric, and may involve small and large joints, the cervical spine, and the temporomandibular joint. The ocular finding tends to be chronic anterior uveitis and occurs in 10% of patients, most commonly in those patients with fewer affected joints.116
Treatment
The goal of treatment for JIA is to control the clinical symptoms of the disease and to limit joint deformity using the most conservative and safest therapy available.116 Initial therapy typically begins with either aspirin or NSAIDs, such as ibuprofen or naproxen. The NSAID dose is dependent on the child’s age and weight. Because of the potential association between aspirin use and Reye syndrome, any aspirin or NSAID should be discontinued in a sick child who potentially has influenza or varicella. Systemic corticosteroids generally are avoided, except in severe systemic disease because of the potential for growth retardation with long-term use. If the patient fails to respond to NSAIDs alone, methotrexate typically is used because it is well-tolerated, has been shown to be effective in the treatment of JIA in a randomized clinical trial, and there is extensive experience with the drug over long-term follow-up.126,127,128 Methotrexate also has been found to be effective in the treatment of JIA-associated uveitis.129,130,131 Other immunosuppressive drug therapies, including cyclosporine and azathioprine, have been used to treat JIA that is refractory to treatment with MTX.116 In cases with persistent inflammation on MTX therapy, biologic therapy is often added in the form of etanercept or adalimumab.116,132,133 However, other biologics have been studied in patients with JIA including infliximab and abatacept, which have shown promising results.116 Patients with polyarthritis (either diagnosed on initial presentation or with the extended oligoarticular form of JIA) are the ones who most commonly need the addition of biologic therapy to control inflammation.
Ocular Manifestations
Approximately 12% to 17% of children with any type of JIA have uveitis.134,135,136 A population-based study performed in Finland reported the mean annual incidence and prevalence rates for JIA-associated uveitis as 0.2 and 2.4 cases per 100,000 population, respectively.137 The ocular manifestations of JIA are two different types of anterior uveitis. Acute anterior uveitis is seen in the HLA-B27–associated subgroups and is similar to that seen in adult HLA-B27–associated disease. Chronic anterior uveitis is seen primarily in the ANA-positive oligoarticular subgroup where frequencies have been reported to range from 20% to 56%.116,121,138,139,140 Although traditionally it has been thought that these cases occur primarily in girls, a recent retrospective study of 90 children with JIA found no gender difference in the risk of developing uveitis.134 The chronic anterior uveitis tends to be asymptomatic or minimally symptomatic; therefore, it is recommended that these patients be evaluated every 3 months for screening in an effort to detect the uveitis early.141 Two thirds of patients have bilateral disease, and in patients with unilateral disease, most will develop uveitis in the fellow eye within 1 year of presentation.116 The chronic uveitis typically requires chronic therapy. Severe visual disability may develop. Band keratopathy and cataracts may be seen in up to one third of patients and are the most frequent causes of decreased vision.142 Secondary glaucoma occurs in 10% to 20% of patients with JIA-associated uveitis and is a poor prognostic finding.136,138,139,142,143 Posterior synechiae are common and have been shown to be associated with poorer visual acuity.142,144 Other complications include macular edema in 8% to 10% of cases and phthisis in 4% to 10% of patients. Although blindness developed in 40% of patients reported in early series, more recent series have reported a better prognosis.140,145,146,147 One study from Finland reported on 426 incident cases of JIA diagnosed between 1989 and 1996, of whom 24% developed uveitis. In this series only 3% of those with uveitis were reported to have a best-corrected visual acuity of 20/60 or worse. The improved prognosis appears to be because of earlier detection, better treatment, and better surgical management of the complications. Although pars plana lensectomy with vitrectomy and aphakic correction traditionally had been considered the best method of cataract surgery in these patients,138,148 more recent case series have suggested that intraocular lenses can be placed in these patients with good visual outcome.148,149
Treatment for the uveitis associated with JIA depends on the type of uveitis. Acute anterior uveitis typically can be controlled with topical corticosteroid eye drops such as prednisolone acetate 1%. The medication is instituted at one drop every 1 to 2 hours while awake until the uveitis is controlled and then tapered slowly and discontinued. Milder forms of chronic uveitis also may be controlled with topical corticosteroids, although the medication may be required indefinitely to keep the uveitis quiet. The dose of topical corticosteroid is tapered to the lowest dose in which the uveitis remains quiet. In cases of more severe uveitis, methotrexate or cyclosporine may be used.129,130,131 Immunosuppressive drug therapy has been shown to reduce the incidence of vision loss and ocular complications (i.e., hypotony and formation of epiretinal membrane) in patients with JIA uveitis.144 Systemic corticosteroids are given in cases of severe, sight-threatening uveitis and are used in conjunction with a steroid-sparing agent such as methotrexate. The systemic corticosteroids are tapered and discontinued as quickly as possible r to avoid side effects such as growth retardation.
Familial Juvenile Systemic Granulomatosis
Familial juvenile systemic granulomatosis, also known as Jabs syndrome, Blau syndrome, and autosomal dominant granulomatous disease of childhood, is an uncommon genetic disease with polyarthritis and uveitis that often is misdiagnosed as JIA or sarcoidosis.150,151,152,153,154 The disease is inherited in an autosomal dominant fashion, and the joint disease is a granulomatous, deforming, nonerosive polysynovitis. Other more variable features of the syndrome include rash, cranial nerve palsies, and vasculopathy.150,151,152,153,154 The syndrome is caused by mutations in the CARD15 gene, which is involved in apoptosis and is preferentially expressed on monocytes.155 The CARD15 gene also has been linked to Crohn disease, but the mutations for the two disorders are in different segments of the gene.155
Children with this syndrome often are misdiagnosed as having either JIA or sarcoidosis. Familial juvenile systemic granulomatosis may be distinguished from JIA by the presence of polyarthritis on presentation, the absence of ANA, and the pattern of inheritance. Although patients with familial juvenile systemic granulomatosis may have a chronic anterior uveitis similar to JIA, these patients also may have multifocal choroiditis, which is not seen in patients with JIA.153 Patients with JIA who have chronic uveitis tend to have oligoarticular arthritis and positive ANA antibodies. Familial juvenile systemic granulomatosis differs from sarcoidosis by the pattern of arthritis, the absence of pulmonary involvement and adenopathy, and the inheritance pattern.153
Ocular Manifestations
The uveitis associated with familial juvenile systemic granulomatosis may be a chronic anterior uveitis or a chronic panuveitis with multifocal choroiditis. Patients may require aggressive medical therapy to control the uveitis, sometimes including immunosuppressive drugs. Ocular complications including cataract and macular edema are common.2
Systemic Lupus Erythematosus
Etiology and Pathogenesis
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease. Although the etiology of SLE is unknown, the association of SLE with HLA-DR2, HLA-DR3, and the association of lupus nephritis with Fcy receptors IIA and IIIA located on chromosome 1 all suggest a genetic predisposition.156,157 Pathogenetically, SLE is characterized by B-cell hyperactivity, polyclonal B-cell activation, hypergammaglobulinemia, autoantibody formation, and abnormal T cells.156 Autoantibodies seen in patients with SLE include ANA, antibodies to DNA, both single-stranded DNA (anti-ssDNA) and double-stranded or native DNA (anti-dsDNA), and antibodies to cytoplasmic components, such as anti-Sm, anti-Ro (SSA), and anti-La (SSB).156 Studies have demonstrated multiple defects in T-cell signaling pathways in patients with SLE that are thought to lead to autoreactivity and hyperactivated T-cell lymphocytes.158,159,160 SLE classically has been regarded as an immune complex disease, in which circulating immune complexes are deposited in tissue and incite an inflammatory response, which may lead to end organ damage. Immunoregulatory abnormalities in SLE lead to an inadequate clearing of immune complexes and further hyperactivation of both B cells and T cells, causing further inflammation and tissue damage.156
Clinical Features
The incidence of SLE has been estimated as 2 to 9 cases per 100,000 population per year.161 SLE is a multisystem disease that may affect almost any organ system.161,162,163,164,165,166 Because of the multisystem nature of the disease, criteria for the diagnosis have been established and are outlined in Table 26-4.167 These criteria are for use in clinical investigations and may not always be applicable in patient management. SLE has a chronic course marked by periodic exacerbations of varying severity.161 Cutaneous disease occurs in approximately 85% of patients with SLE. The most classic manifestation is the characteristic butterfly rash across the nose and cheeks, also known as a malar rash. Other cutaneous lesions include discoid lupus erythematosus, vasculitic skin lesions such as cutaneous ulcers or splinter hemorrhages, purpuric skin lesions, alopecia, and livedo reticularis, a reticular purplish rash seen most commonly on the legs. Less common skin lesions include a maculopapular eruption, lupus profundus, bullous skin lesions, and urticarial skin lesions. Mucosal lesions occur in 30% to 40% of patients with SLE and characteristically are painless oral ulcers. Photosensitivity is a common feature of the skin lesions in SLE affecting up to 70% of patients, and patients may be exquisitely sun sensitive.161,162,163,164,166 The Raynaud phenomenon occurs in approximately 20% of patients with SLE. Cardiac disease includes pericarditis, occasionally myocarditis, and Libman-Sacks endocarditis. Libman-Sacks endocarditis was common in old autopsy series but now is unusual. Pleuropulmonary lesions include pleurisy and, less commonly, pneumonitis. Adenopathy and hepatosplenomegaly can be seen in 50% of patients with SLE.161,162,163,164,166 Involvement of the central nervous system (CNS) by lupus occurs in 35% of patients with SLE.165,166 CNS lupus may present as seizures, an organic brain syndrome, or psychosis.165 Transverse myelitis is an uncommon manifestation, occurring in only 4% of patients with SLE, but often is seen in association with optic neuritis. Peripheral nervous system involvement with a peripheral neuropathy and cranial nerve palsy is less common.
Table 26-4. The 1982 Revised Criteria for the Diagnosis of Systemic Lupus Erythematosus | ||
|---|---|---|
|
At some point during the course of their SLE, 80% to 85% of patients with SLE have articular disease. Articular involvement includes polyarthralgias and a nondeforming migratory polyarthritis. Cutaneous nodules, myalgias, and myositis are less common. Systemic features occur in more than 80% of patients with lupus and include fatigue, fever, and weight loss. Renal disease is present in approximately 50% of these patients. Lupus nephritis clinically presents as either proteinuria with a nephritic picture or as glomerulonephritis with active urinary sediment. Lupus nephritis is a major cause of morbidity and mortality in SLE.161,162,163,164,166 Patients with SLE often have a mild chronic anemia, known as anemia of chronic disease. However, they may also develop an autoimmune hemolytic anemia. Leukopenia, in particular lymphopenia, is a characteristic feature. Thrombocytopenia occurs in approximately one third of patients.161,162,163,164,166
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


