Ocular Manifestations of Gastrointestinal Diseases
Anil K.V. Aralikatti
Anju Kadyan
Uday Bhatt
Dipali Aralikatti
Damian O’Neill
Ocular changes are associated with many developmental and metabolic conditions affecting the liver, pancreas, and intestine. The changes may also be secondary effects of gastrointestinal (GI) diseases such as nutritional deficiencies or metastases. These ocular features have diagnostic, therapeutic, and prognostic implications. In this chapter we deal topographically with the ocular findings in a range of GI diseases.
Eyelid Manifestations
Xanthelasma is a feature of hyperlipidemia and prolonged cholestasis. Xanthelasma and xanthomas are seen in patients with primary biliary cirrhosis although they are seen less frequently now.1
Primary biliary cirrhosis (PBC) is a chronic, slowly progressive, intrahepatic autoimmune cholestatic liver disease that eventually leads to liver failure. The diagnosis is established by the triad of antimitochondrial antibody, cholestatic indices, and liver histology. Although antimitochondrial antibody is present in more than 90% of cases, antibody titers and profiles do not show any clinical correlation.2 Antimitochondrial antibody–negative patients with cholestatic disease need careful evaluation with cholangiography and liver biopsy. Ursodeoxycholic acid improves liver biochemistry and delays histologic progression in early disease. Many of the complications of the disease such as pruritus and osteoporosis are amenable to treatment.3 Liver transplantation is the only effective treatment for end stage PBC, showing one of the best survival outcomes of transplantation for any liver disease.4
Florid xanthelasma (yellow lid sign; Fig. 30-1) with orbital inflammation is reported in Wegener granulomatosis, a systemic necrotizing vasculitis.5 Wegener is a multisystem disease involving upper airways, lung, kidney, and occasionally the GI tract in the form of esophageal vasculitis, colitis, GI hemorrhage, and bowel perforation.6
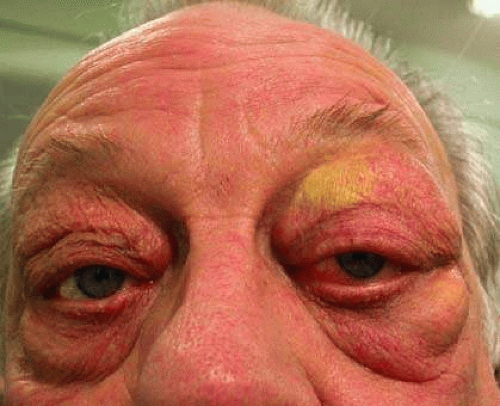 Fig. 30-1. Xanthelasma in Wegener granulomatosis. (Courtesy of S. Sandramouli.) |
Orbital Manifestations
Frontal bossing with shallow nasal bridge is a feature of many of the lysosomal storage diseases (mucopolysaccharidoses and mucolipidoses), whereas hypertelorism is sometimes seen in patients with generalized gangliosidosis.9 Congenital intrahepatic cholestasis is frequently accompanied by hypertelorism.10
Cavernous hemangiomas are the most common benign tumors of the orbit in adults. Bilateral, multifocal lesions may be complicated by life-threatening GI bleeds. These have been reported in the blue rubber bleb nevus syndrome (multiple distinctive skin and GI tract hemangiomas), Maffucci syndrome (visceral hemangiomas in the esophagus, ileum, and oral mucosa with bony lesions, enchondromas that affect small bones of hands and feet), Rendu-Osler-Weber syndrome (mucocutaneous and visceral telangiectasia usually in the liver and upper GI tract), and diffuse neonatal hemangiomatosis.11
Conjunctival Manifestations
Jaundice
Hyperbilirubinemia causes a yellow discoloration of tissues containing elastin such as skin and mucosa appreciable when bilirubin concentrations exceed 34μ mol/L (2 mg/100 dL). The bilirubin deposition is usually in the conjunctiva, although peripheral corneal stromal deposits have also been reported.12
Leptospirosis is a zoonosis acquired when humans are exposed to infected urine of carrier mammals during activities such as agriculture and water sports.13 The biphasic illness manifests in the septicemia phase with fever, headache, myalgia, conjunctival chemosis, and subconjunctival hemorrhage. Severe illness is characterized by jaundice, renal failure, and hemorrhagic manifestations including retinal hemorrhages and papillitis. Jaundice with circumcorneal congestion is regarded as a pathognomonic sign of severe leptospirosis. The immune phase characterized by nongranulomatous panuveitis, vasculitis, and interstitial keratitis follows a prolonged symptom-free period (2 days to 4 years). Other ocular complications include glaucoma and cataract. Rapid maturation of cataract and spontaneous absorption of the lens is a rare but unique association.14 In the tropics, it is one of the most common causes of hypopyon uveitis. Recommended treatment includes doxycycline for prophylaxis and mild disease, and penicillin G, ampicillin, and ceftriaxone for severe disease. Treatment provides greatest benefit when initiated early in disease, but it may not prevent immune-related ocular disease.13,15
Keratoconjunctivitis Sicca
Keratoconjunctivitis sicca affects many patients with autoimmune diseases. More than half of the patients with PBC have at least one associated autoimmune disease, the most common being sicca syndrome (25%–50%).1 Disease concurrence is significantly associated with the presence of anticentromere antibody.16
Keratoconjunctivitis sicca is the most frequent ocular manifestation of scleroderma, a chronic multisystem disorder characterized by immune activation, vasculopathy, and widespread fibrosis.17 GI involvement includes gastroesophageal reflux, dysphagia, malabsorption syndrome, intestinal pseudo-obstruction, constipation, and fecal incontinence. The dry eye features are aggravated by progressive lid stiffness and lagophthalmos. The conjunctival vessels show telangiectasia with shallowing of the fornices due to subepithelial fibrosis.
An altered immune system or direct infection of the lacrimal gland may be responsible for a strong association of Sjögren syndrome and hepatitis C virus (HCV).18,19 HCV-related Sjögren syndrome varies in clinical and serologic testing from primary Sjögren syndrome. The HCV patients are older, have less parotidomegaly, but have a higher prevalence of liver involvement, antimitochondrial antibodies, cryoglobulinemia, and hypocomplementemia.20
Conjunctival Phlyctenulosis
Phlyctenulosis of the conjunctiva is a delayed hypersensitivity reaction to bacterial, fungal, and parasitic antigens. Common associated intestinal parasites are Hymenolepis nana and Ascaris lumbricoides. Treatment with topical steroids and eradication of the intestinal parasite can lead to resolution of recurrent ocular surface inflammation.21
Conjunctival Anomalies in Lysosomal Storage Diseases
Minor tear and conjunctival anomalies are common in lysosomal storage diseases. Bulbar conjunctival microaneurysms are frequently seen in the mucolipidoses, and pingueculae are a feature of adult Gaucher disease.22,23 Conjunctival cytologic studies are useful in the diagnosis of mucolipidosis type IV.24
Corneal Manifestations
Asymptomatic peripheral corneal opacification is found in several chronic hepatobiliary disorders, whereas progressive central corneal opacification is a feature of many of the mucolipidoses and mucopolysaccharidoses. Peripheral ulcerative keratitis may be the first sign of systemic necrotizing vasculitis in Wegener granulomatosis (Fig. 30-2) and polyarteritis nodosa.25 Nonhealing corneal ulcers, especially in patients with malabsorption, can be related to vitamin A deficiency and respond well to supplementary therapy.26
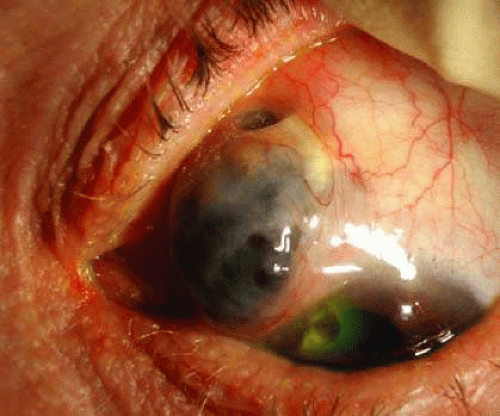 Fig. 30-2. Peripheral ulcerative keratitis with scleritis in Wegener granulomatosis. (Courtesy of S. Pagliarini.) |
The Cornea in Wilson Disease
Wilson disease is an autosomal recessive disorder with impaired hepatic excretion of copper, which consequently accumulates in the liver, kidney, cornea, and other end organs. Liver damage typically presents in a child as cirrhosis, jaundice, and signs of portal hypertension, including ascites, splenomegaly, and esophageal varices. Central nervous system (CNS) damage chiefly affects the basal ganglia, causing incoordination, spasticity, rigidity, tremor, or dysarthria. Psychiatric disturbances are common.27
The distinctive ocular sign of Wilson disease is the Kayser-Fleischer ring (Fig. 30-3). This peripheral corneal copper deposition is at the level of the Descemet membrane and may appear green, brown, or red. It is not pathognomonic of Wilson disease since similar rings may occasionally be seen in other hepatic conditions.28 The ring is usually found in patients with neurologic manifestations of Wilson disease.29 Anterior and posterior subcapsular copper deposition may lead to the pathognomonic sunflower cataract formation (Fig. 30-4).30 The ocular features may be present long before hepatic or neurologic manifestations.30,31
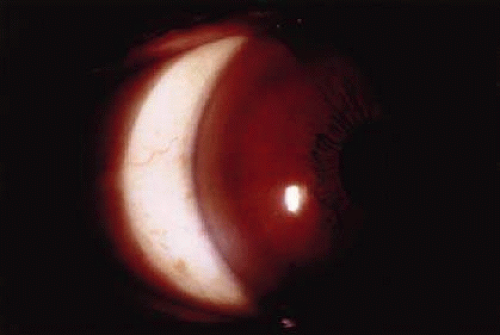 Fig. 30-3. Kayser-Fleischer ring in Wilson disease. (Courtesy of P. McDonnell.) |
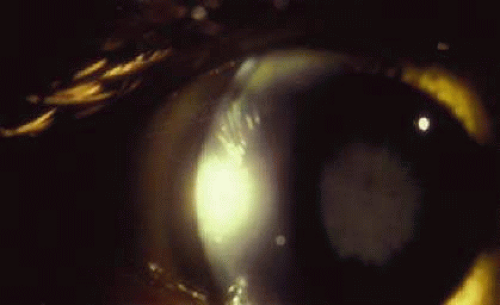 Fig. 30-4. Sunflower cataract in Wilson disease. (Courtesy of D. O’Neill.) |
The treatment of Wilson disease includes lifelong administration of chelating agents (d-penicillamine, trientine) or zinc. Kayser-Fleischer rings regress after systemic therapy, but this does not correlate with clinical improvement.32,33 Advanced liver disease and treatment-resistant cases require liver transplantation.29,34
The gene for Wilson disease, ATP7B, is located on chromosome 13. Genetic mutation analysis is a reliable tool for screening first-degree relatives of an index case with known mutation.34,35 Hence it is possible to identify and treat other affected siblings at an early age before they develop overt biochemical changes with irreversible hepatic or neurologic damage.
Peripheral Corneal Rings in Non-Wilsonian Chronic Liver Disease
Peripheral Corneal Changes in Alagille Syndrome
Alagille syndrome is a rare autosomal dominant disorder characterized by paucity of interlobular bile ducts and five major clinical features: neonatal chronic cholestasis, characteristic facies, cardiovascular abnormalities, vertebral arch defects, and posterior embryotoxon.38 It is caused by mutations in the Jagged 1 gene on chromosome 20p12.39 Other known mutations involve the gene for the Notch 2 receptor.40 The distinctive facial appearance (prominent forehead, deep-set eyes, hypertelorism, straight nose, and small pointed chin) is an important clinical sign in the diagnosis of Alagille syndrome.10
Posterior embryotoxon occurs in more than 80% of patients.10 It is, however, found in 10% of the general population,38 and it is therefore possible that posterior embryotoxon may be a coincidental finding in a jaundiced newborn rather than a manifestation of Alagille syndrome. It is important not to make an erroneous diagnosis of Alagille syndrome and overlook treatable extrahepatic biliary atresia.
Other common ocular abnormalities of Alagille syndrome include microcornea, iris abnormalities, diffuse fundus hypopigmentation, retinal pigment granularity, optic disc anomalies, and choroidal folds.41 Optic disc drusen is common, and its presence is useful in the diagnosis of the condition.42 Some children have progressive visual loss associated with idiopathic intracranial hypertension and require monitoring with annual funduscopy.43 Despite advances in genetics, the correlation between genotypes and phenotypes remains incompletely defined.44
Liver transplantation has been used to successfully treat patients with end stage liver failure, portal hypertension, severe pruritus, and xanthomatosis.38
Corneal Changes in Inflammatory Bowel Disease
Corneal changes are seen in 0.5% of patients with inflammatory bowel disease (IBD). It is characterized by peripheral subepithelial nodular infiltrates that coalesce, leading to peripheral corneal opacities. There may be secondary pannus and progressive astigmatism with peripheral corneal thinning due to vasculitis-induced collagenolysis.45 Some patients may have recurrent nonspecific follicular conjunctivitis or granulomatous conjunctivitis.46
Corneal Arcus
Corneal arcus (Fig. 30-5) is caused by extracellular lipid deposition and presents as a yellow-white band in the peripheral cornea separated from the limbus by a narrow clear zone. It has a strong association with age and was considered to predict cardiovascular disease and coronary heart disease.47 However, after adjusting for age, it may not be an independent risk factor for predicting vascular complications.48
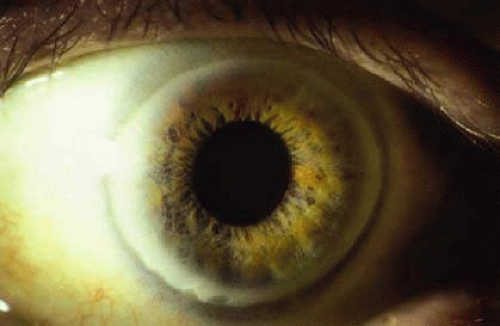 Fig. 30-5. Corneal arcus. (Courtesy of D. O’Neill.) |
Many primary hyperlipidemias are due to abnormalities of hepatic lipid metabolism. Premature corneal arcus is frequently seen in association with familial hypercholesterolemia, and has also been reported in type III, type IV, and type V hyperlipoproteinemia.49,50 Winder has surmised that the prevalence of corneal arcus in familial hypercholesterolemia is related to the age of the patient and the duration of hyperlipidemia rather than to the severity or type of hyperlipidemia.50
Familial hypercholesterolemia is an autosomal dominant defect of low-density lipoprotein receptors due to a mutation in chromosome 19.51 The clinical criteria for the diagnosis of this condition include elevated cholesterol levels, family history of hypercholesterolemia, premature cardiovascular disease, presence of tendon xanthomas, and corneal arcus before 45 years of age.51 In homozygotes, corneal arcus is present in childhood whereas in heterozygotes the arcus usually develops after 30 years of age.52
Corneal arcus is a prominent and early finding in type III hyperlipoproteinemia (familial dysbetalipoproteinemia). Affected patients have xanthomas (particularly of the palmar creases: xanthoma striatum) and premature atherosclerosis. This condition is caused by a mutation of the apo E gene, which leads to impaired clearance of remnant lipoproteins by the liver.53
Lecithin cholesterol acyl transferase (LCAT) deficiency is a rare autosomal recessive metabolic abnormality localized to chromosome 16q22.54 Very high concentrations of unesterified cholesterol are found because of almost total absence of lysolecithin and absent or reduced LCAT function. Premature corneal arcus occurs before the other systemic features, which include atherosclerosis, proteinuria, renal failure, and hemolytic anemia.55
Fish-eye disease is a defect of lipid metabolism believed to be related to LCAT deficiency.56 The peculiar ocular finding is of profound diffuse corneal stromal lipid infiltration and opacification believed to be similar to the corneal opacification found in boiled fish.57 The clinical abnormality is confined to the cornea in this condition, and progressive disabling opacification is apparent from the second decade onward.
Tangier disease is a rare autosomal recessive disorder associated with deficiency of high-density lipoproteins. It is caused by mutations in the gene encoding ATP-binding cassette transporter 1.58 Cholesterol esters accumulate in lymph nodes, liver, spleen, intestinal tract, and peripheral nerves. There is visible enlargement of the tonsils, which take a characteristic orange-yellow color. The most common ocular abnormality is corneal opacification, usually apparent only on slitlamp examination.59 Strabismus, ectropion, and retinal pigment epithelial (RPE) abnormalities have also been reported in this condition.52
The association of corneal arcus with coronary heart disease and cardiovascular disease in the Lipid Research Clinics Mortality Follow-up Study yielded definitive advice on which patients with corneal arcus should be investigated for hyperlipidemia. This study followed more than 10,000 patients and concluded that all patients younger than the age of 50 years with corneal arcus should have a detailed evaluation of their coronary vascular disease risk factors, including measurement of serum lipids.60
Central Corneal Opacification and Hepatomegaly: Mucolipidoses and Mucopolysaccharidoses
Mucopolysaccharidoses (MPSs) and mucolipidoses (MLSs) are inherited lysosomal enzyme deficiencies. The enzyme deficiency leads to the accumulation of mucopolysaccharides (glycosaminoglycans) or mucolipids that the deficient enzyme would normally metabolize. The enzyme deficiencies are expressed at the level of the bone marrow stem cells, and thus these disorders fall into the relatively small number of inborn errors of metabolism that are amenable to correction by allogenic bone marrow transplantation.61 They are all autosomal recessive with the exception of MPS type II (Hunter syndrome), which is X-linked recessive.
The resultant accumulation of intracellular material causes hepatosplenomegaly, mental retardation, and skin, skeletal, and ocular changes with varying prevalence, depending on the subtype of disease involved and the age at which the patient is examined. There are seven types of MPSs with a broad continuum of severity and range of manifestations.62
Increased urinary excretion of glycosaminoglycan is a feature of the MPSs but not a feature of the MLSs. Further subclassification depends on the patient’s phenotype and family pedigree and identification of the deficient enzyme from cultured skin fibroblasts, cultured amniotic cells, peripheral blood leukocytes, or conjunctival biopsy (although the conjunctiva is usually clinically normal in these groups of conditions).63
The prevalence of clinically apparent corneal opacification depends on the type of disease, age of the patient, the method of examination, and the experience of the examining physician.
Severe corneal clouding within the first few years of life is typical of MPS type I H (Hurler syndrome) and type VI (Maroteaux-Lamy syndrome), whereas the onset of corneal opacification may occur from birth to age 20 in MPS type I S (Scheie syndrome).64,65,66
Corneal opacification is found much less frequently in other MPSs. Approximately 10% of patients with MPS type IV (Morquio syndrome) develop corneal opacities after age 10 years, and corneal opacities are also seen in a few patients who have the milder phenotypes of MPS type II and rarely MPS type III (Sanfilippo syndrome).63,65,67
Corneal opacification is present in early childhood in almost all cases of type IV MLS.68 Mild opacification, usually evident by age 10 years, is found in all cases of type III MLS.69 Mild corneal opacification is a feature of 40% of cases of type II MLS and less than 20% of type I MLS and generalized gangliosidosis.70,71,72
Episodic ocular pain is an important symptom in MLS type IV. It is caused by corneal epithelial cytoplasmic accumulation of abnormal material with subsequent corneal surface irregularities.73
Severe corneal opacities can be treated by penetrating keratoplasty, although the visual prognosis may be limited by several factors, including retinopathy, glaucoma, and optic nerve disease, as well as the risk of cataract and recurrence of opacification in the corneal graft.63,74
Successful early bone marrow transplantation for systemic MPSs has shown to improve systemic health and reduce corneal opacification in some patients.75 Recent trials of enzyme replacement therapy for MPSs have been encouraging, and this treatment has become commercially available for MPS type I.62,76,77
Glaucoma
Helicobacter pylori, a Gram-negative bacterium that colonizes the gastric mucosa, has been associated with upper GI diseases as well as extraintestinal diseases.78 These manifestations may be related to autoimmunity; and this has led to research for a possible link between H. pylori and glaucoma. Increased levels of IgG antibodies have been demonstrated in the aqueous humor of patients with primary open-angle glaucoma. Eradication of H. pylori has been proposed to have a beneficial effect on intraocular pressure, visual field parameters, and glaucoma progression.79,80,81,82
Histamine receptor antagonists (cimetidine and ranitidine) used in peptic ulcer have a weak anticholinergic effect and may induce mydriasis with subsequent angle closure in anatomically predisposed eyes.83 Conversely, antiglaucoma medications can affect the GI system: Cholinergic agonists can cause salivation, diarrhea, and acid secretion; systemic carbonic anhydrase inhibitors can alter stomach pH and cause diarrhea. Topical prostaglandin analogues can induce gastric crisis with esophageal spasm, gastric reflux, and abdominal cramps by stimulation of the GI smooth muscle.84 Although rare, these effects need to be considered in patients with GI symptoms needing concomitant antiglaucoma medication.
The MPSs may be associated with open-angle or angle-closure glaucoma. Accumulation of glycosaminoglycans can result in narrowing of the angle structures and obstruction of trabecular outflow.85,86,87,88 Intraocular pressure measurement may be affected by corneal thickening. Assessment of the optic disc, drainage angle, and peripheral iridotomy may be hampered by corneal opacification. The young age and intellectual impairment of the patient makes visual field testing difficult or unreliable. Glaucoma and ocular hypertension may also present following bone marrow transplantation for MPSs.61,75
Lens Manifestations
Prolonged systemic adrenocorticosteroid therapy (such as in the treatment of chronic IBD, inflammatory eye disease, or after liver transplantation) may be complicated by secondary cataract formation. Liver disease with cataract formation is a feature of galactosemia, Zellweger (cerebrohepatorenal) syndrome, and cerebrotendinous xanthomatosis.
Galactosemia
Classical galactosemia is an autosomal recessive defect of galactose metabolism due to a deficiency of the enzyme galactose-1-phosphate uridyl transferase. The resultant accumulation of galactose leads to hepatocellular damage (with cirrhosis and ascites), renal tubular acidosis (with aminoaciduria), mental retardation, and cataract formation. Severe cases present in infancy as diarrhea, vomiting, and hepatomegaly, and then cataract formation after commencing milk feeding. Recently, coagulopathy-associated vitreous hemorrhage has been reported in cases with severe galactosemia.89 The diagnosis of galactosemia is confirmed by demonstrating deficient erythrocyte galactose-1-phosphate uridyl transferase enzyme activity.
Routine neonatal metabolic screening for galactosemia is undertaken in many countries. Early diagnosis and galactose-restricted diet reduces acute morbidity and mortality and prevents cataract formation and progression.90,91 However, it does not prevent the long-term complications of the disease such as retarded mental development, verbal dyspraxia, motor abnormalities, and hypergonadotrophic hypergonadism.90,92
Zellweger Syndrome
Peroxisomes are subcellular organelles that perform various important anabolic and catabolic functions, including the degradation of very-long-chain fatty acids. Disorders of peroxisome biogenesis include Zellweger syndrome and neonatal adrenoleukodystrophy.94,95,96 Cataracts are a feature of Zellweger syndrome (in both homozygotes and heterozygotes) whereas the anterior segments are usually normal in neonatal adrenoleukodystrophy.97 Both of these conditions are discussed in greater detail later in the section on retinal manifestations.
Cerebrotendinous Xanthomatosis
Cerebrotendinous xanthomatosis is a rare autosomal recessive disorder of bile acid synthesis due to deficiency of the mitochondrial sterol 27-hydroxylase. The disease is characterized by tendon xanthomas, juvenile cataracts, neurologic abnormalities, and premature atherosclerosis. Bilateral juvenile cataracts with chronic diarrhea represent the earliest clinical manifestations of cerebrotendinous xanthomatosis.98 Optic neuropathy has also been reported.99 The clinical diagnosis is confirmed by the demonstration of raised plasma cholestanol concentrations. Oral chenodeoxycholic acid replacement therapy arrests the progression of the disease and may reverse some of the manifestations.
Retinal Manifestations
Many diseases of the GI system affect the retina. Although many of these have a causal relationship, others are linked and manifested together due to being close to each other on the chromosome locus.
Retinopathy in Systemic Mucopolysaccharidoses
A variable degree of retinopathy occurs in many types of MPSs. MPS I H (Hurler), MPS I H-S (Hurler-Scheie), MPS I S (Scheie), MPS II (Hunter), and MPS IV (Morquio) show mild to moderate retinopathy. Moderate to severe retinopathy is a common feature of MPS III (Sanfilippo). The retinopathy has an insidious onset but is often masked by corneal opacification. Clinical signs include arteriolar narrowing, indistinct foveal reflex and pigmentation, RPE atrophy, and later bone spicules, pallor of the optic disc, and associated visual field constriction.85 Electroretinography (ERG) changes occur in many of the MPS types, with rod function more severely affected than that of cones.100
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


