Ocular Manifestations of Autosomal Dominant Systemic Conditions
Majida Gaffar
Steven E. Rubin
Leonard B. Nelson
This chapter reviews those autosomal dominant conditions that have associated eye findings. Familiarity with these ocular manifestations places the ophthalmologist in a unique position of helping to provide a systemic diagnosis in affected patients.
AUTOSOMAL DOMINANT INHERITANCE
Heritable conditions that follow Mendelian genetics are autosomal dominant, autosomal recessive, X-linked dominant, or X-linked recessive. Characteristics of the nonautosomal dominant modes of inheritance are reviewed elsewhere.
An autosomal dominant trait must exhibit certain characteristics. There must be vertical transmission (i.e., the trait appears in consecutive generations). A heterozygote exhibits the trait in some way. A parent of either gender with the trait passes it on to 50% of his or her children independent of the gender of the children. (The gender incidence of this trait would then be equal.) Children of unaffected parents are affected only by mutation.
Penetrance and expressivity are two parameters that have a bearing on autosomal dominantly inherited disease. Penetrance is the ability of a genotype to be phenotypically detected. If a subject is known to have an autosomal dominant genotype and yet has no detectable phenotype, that gene is impenetrant. Penetrance is binary; an autosomal dominant gene is either penetrant or impenetrant. Expressivity refers to the degree of variability that a penetrant autosomal dominant trait can have. For example, some affected individuals with neurofibromatosis may have few cutaneous neurofibromas, whereas others are severely disfigured.
Autosomal dominant diseases can also have seemingly unrelated manifestations in different organ systems and tissues, although only one gene is present. This is called pleiotropy.
MULTISYSTEM DISEASE
Marfan Syndrome
A description of what we now call Marfan syndrome was probably first made by Williams, an ophthalmologist, in 1895, who did not include ectopia lentis as a manifestation. Ironically, it was a pediatrician who first published that association in 1914. Marfan’s description of a 5½-year-old child with long, thin extremities, published in 1896, was more widely seen than Williams’ the year before, and hence this syndrome now bears his name.
Marfan syndrome is a connective tissue disorder with an autosomal dominant pattern of inheritance. It has a prevalence of 4 to 6 per 100,000 with no gender, racial, or ethnic predilection. Sporadic cases are often associated with advanced paternal age and are thought to occur in approximately 15% of cases. Advanced technology now allows for prenatal diagnosis with linkage analysis or direct gene sequencing.1
Chromosome 15 (locus 15q21.1) has been identified as harboring the genetic defect for this disorder, which results from a mutation in the fibrillin 1 gene (FBN1).2 Fibrillin is an important component of microfibrils that are necessary for formation of tissues and also necessary to the elastic property of tissues. The abnormal fibrillin leads to structural weakness resulting in the phenotypic appearance of an individual with Marfan syndrome.3
Systemic Manifestations
Marfan syndrome’s nonocular systemic manifestations are found in the skeletal and cardiovascular systems.
Cardiovascular abnormalities are the more significant systemic manifestations. In more than 95% of the cases in which a cause of death can be established, a cardiovascular problem is at fault.4 Dilatation of the aortic root (with or without aortic regurgitation), aortic aneurysm, mitral valve prolapse, and mitral regurgitation are all seen. Pregnancy can exacerbate these cardiac complications, more often in the third trimester. Treatment with β-blockers is now the standard of care to delay or prevent aortic aneurysm and dissection. Verapamil and angiotensin-converting enzyme inhibitors are viable options for patients unable to tolerate β-blockers.5
Abnormalities of the skeletal system include scoliosis, arachnodactyly, anterior chest deformities, dural ectasia (present in 92% of patients), increased arm span to total height ratio, joint laxity, and a high-arched palate. Spontaneous pneumothorax and apical blebs have been described in this disorder as well. Scoliosis is the most disabling skeletal abnormality and can be progressive.3,5
Ocular Manifestations
Ocular manifestations occur in as many as 60% of patients. Of the many ocular abnormalities, by far the most common is ectopia lentis, occurring in 50% to 80% of affected individuals (Fig. 58.1). It is usually bilateral, symmetric, and nonprogressive. It may be subtle and detectable only by observing phacodonesis or iridodonesis, sometimes visible by gonioscopy. Lens dislocation is characteristically, but not always, superotemporal. There can also be posterior displacement, leaving a gap between the lens and the iris pigment epithelium. It is thought to occur in utero and therefore can be identified early in life. In approximately 10% of individuals, glaucoma can be associated with ectopia lentis.1
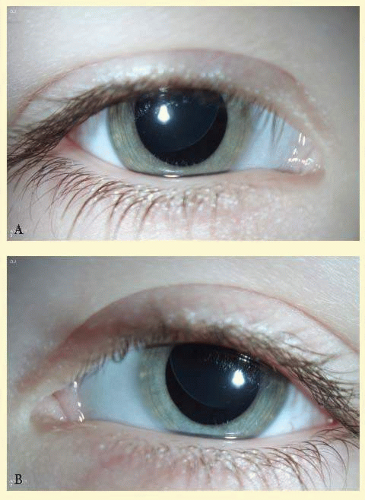 FIG. 58.1 Bilateral ectopia lentis (superotemporal dislocation is most common) in a child with Marfan syndrome; top—right eye, bottom—left eye. |
Patients with Marfan syndrome may have poor vision. This often results from delayed and inadequate correction of the extremes of refractive error (especially moderateto-high myopia).6 These and other factors that compromise visual input may be responsible for the increased incidence of strabismus seen in patients with Marfan syndrome.7
In Marfan syndrome, an increased axial length may also be seen. A slightly higher than normal axial length is common in patients with Marfan syndrome who have no ectopia lentis or retinal detachment. However, the axial length is higher in those with ectopia lentis and is even higher in those with retinal detachment8; retinal detachments occur in 5% to 11% of patients with Marfan syndrome and increase to 40% if there is ectopia lentis.2 Other retinal associations include lattice degeneration and atrophic holes.
Abnormalities may also be seen in the uveal tract. Choroidal thinning has been described in myopic eyes. The iris morphology is often striking. There can be a complete absence or marked reduction in the number of furrows or crypts, giving the anterior iris surface a smooth, velvety appearance (Fig. 58.2). Hypopigmentation of the iris pigment epithelium has been described, which probably corresponds to the transillumination of the iris base that is seen in 10% of cases.9,10 The pupils are typically small and dilation is often difficult because of atrophy of the dilator muscle fibers.1
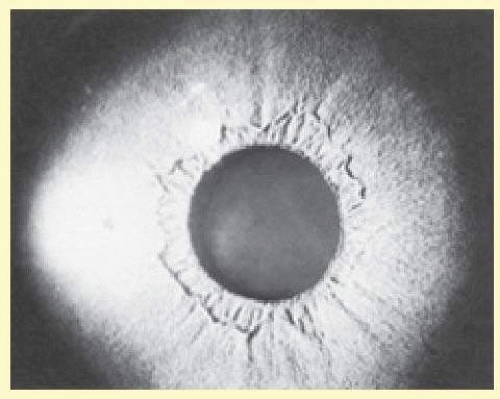 FIG. 58.2 Velvety-smooth iris in a patient with Marfan syndrome. |
The cornea can also be affected. Patients with Marfan syndrome can have flatter than average keratometric readings.8,11 Megalocornea12 may also be present. In eyes with ectopia lentis (with or without retinal detachment), keratometric readings may show steepening in the meridian of the lens dislocation.
Other, nonspecific findings have been described in the few ocular histologic studies that have been performed in Marfan syndrome. Separations of individual zonular fibers into their composite filaments6 and angle anomalies have been reported.9,10,13 Angle abnormalities, found in 75% of eyes, are marked by thick trabecular meshwork and dense iris processes.14 Blue sclera, scleral thinning, and lens colobomas have also been identified.2
The Phacomatoses
The phacomatoses are a group of heredofamilial conditions that all have as a hallmark various hamartomas. The term comes from the Greek term for birthmark (mother spot). Newer literature identified the phacomatoses as neurocutaneous disorders.1 We only consider the autosomal dominant disorders in this chapter (neurofibromatosis, tuberous sclerosis, and angiomatosis retinae). Despite the lack of hard evidence for autosomal dominant transmission, encephalotrigeminal angiomatosis (Sturge-Weber syndrome) is also discussed for completeness.
Neurofibromatosis
Manifestations of this condition had been observed for ages before being described by Robert William Smith in 1849. The more classic description was given by the German pathologist Friedrich Daniel von Recklinghausen who accurately described the diverse findings as a single entity in 18821; it is often referred to as von Recklinghausen disease. Probably because of the complex effects on these patients’ personality as a result of the sometimes severe disfigurement, neurofibromatosis maintains a high profile in the lay press15,16,17,18,19 and the literature (e.g., The Elephant Man, a 1979 Broadway production written by Bernard Pomerance, and a 1980 British motion picture, although the correct diagnosis for the patient portrayed in those works was probably Proteus syndrome).20
Neurofibromatosis is a proven autosomal dominant condition with variable penetrance and expressivity. This, too, appears to have no gender, racial, or ethnic predilection. There are two different types, neurofibromatosis 1 (NF1) and neurofibromatosis 2 (NF2). The former affects 1 in 3,500 individuals, whereas the latter has a lower prevalence of 1 in 33,000 to 40,000. The gene for NF1 has been mapped to chromosome 17q11.2, whereas NF2 results from an abnormality or a deletion of the entire gene on chromosome 22. This less common variant denoted the syndrome of bilateral vestibular schwannomas occurring in 85% of patients.1,21 NF2 was first described by J. H. Wishhart in a patient with bilateral acoustic neuromas and multiple meningeal tumors.1 Current diagnostic criteria of NF1 include any of the following two clinical features: (1) six or more café-au-lait spots (Fig. 58.3), (2) axial or inguinal freckling, (3) two or more Lisch nodules, (4) two or more neurofibromas (Fig. 58.4), (5) optic nerve glioma, (6) distinctive osseous lesions, and (7) a first-degree relative with NF1.2 These diagnostic criteria are less sensitive for children. A retrospective study of 1,900 cases conducted in 2000 found that 46% of sporadic cases did not meet these criteria at age 1; at age 8, 97% met the criteria; and by age 20, 100% met the criteria.21
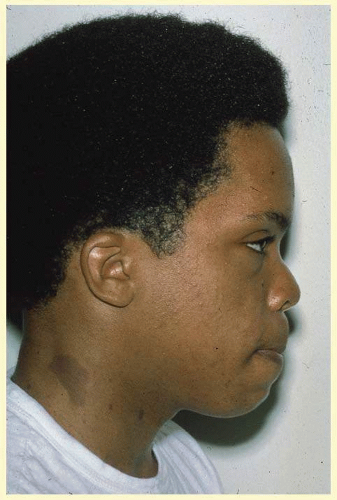 FIG. 58.3 A 13-year-old patient with neurofibromatosis. Note the large café-au-lait spot at the base of the neck. |
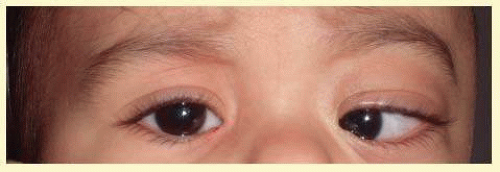 FIG. 58.4 Plexiform neurofibroma of the orbit presenting as progressive proptosis, esotropia, and abduction deficit in a 1-year-old child with neurofibromatosis type I. |
Although clinical findings are primarily neurocutaneous in nature, any organ system can be involved. The cutaneous tumors, which are common and can be extremely disfiguring, take one of three forms. A fibroma molluscum is usually referred to as the common neurofibroma. This hamartoma is the proliferation of peripheral nerve elements at the distal end of a cutaneous nerve. A plexiform neurofibroma of the eyelid (Fig. 58.5) has the appearance of a “bag of worms” and represents a diffuse proliferation of tissue within the nerve sheath. The most marked cutaneous change results from diffuse proliferation outside the sheath, elephantiasis neuromatosa. When a hamartoma develops in a restricted space, a compromise in that nerve’s function can occur.
Neurofibromas may occur anywhere in the CNS. They can produce a neurologic deficit corresponding to the affected nerve. Although these neurofibromas are generally benign, malignant peripheral sheath tumors can occur, the overall risk being about 5%.1 Meningiomas can also be seen with a higher incidence in the general population, especially involving the orbital portion of the optic nerve.22 When they occur in this location, they are more aggressive than their benign counterparts found in other locations.
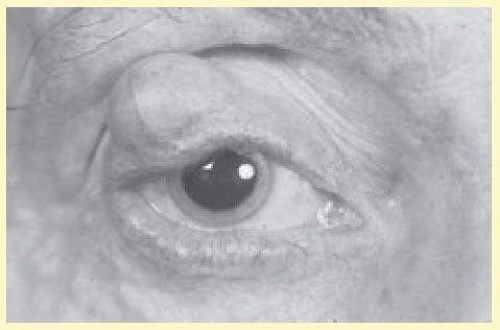 FIG. 58.5 Plexiform neurofibroma in right upper lid of a patient with neurofibromatosis. (Courtesy of David B. Schaffer, MD.) |
Pheochromocytoma has been noted more frequently in patients with neurofibromatosis than in the general population, although it may be difficult to distinguish the clinical features of neurofibromatosis from multiple endocrine neoplasia type IIb, in which pheochromocytomas also occur along with neuromas, café-au-lait spots, and prominent corneal nerves.23 Other tumors, such as rhabdomyosarcoma and liposarcoma, have rarely been reported with this disorder, and this may represent an unrelated association.24
Skeletal deformities, unrelated to the neural tumors, may also occur. Scapular elevation, asymmetries or absences of long bones, misshapen sphenoid or absence of its greater wing, scoliosis, spina bifida, rib fusion, and many others have all been described (Fig. 58.6).24,25,26
Mild mental retardation or learning deficits affect 30% to 40% of affected individuals. This may be caused by mutations disrupting neurofibromin, the protein product of the NF1 gene.1
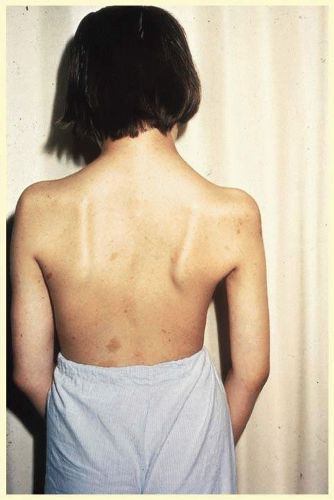 FIG. 58.6 Scoliosis and café-au-lait spots in a patient with neurofibromatosis. (Courtesy of David B. Schaffer, MD.) |
Individuals affected with NF2 often have bilateral acoustic neuromas, which are Schwann cell tumors arising from the vestibular nerves, producing hearing loss or unsteadiness of gait in the second or third decade of life.27 Café-au-lait spots, cutaneous neurofibromas, and plexiform neurofibromas may also be seen in these patients and may even predate the acoustic nerve symptoms.
Ocular Manifestations
Ocular involvement in NF1 involves the cornea, uvea, trabecular meshwork, optic nerve, and retina. Corneal nerves can be thickened; the nerves in the conjunctiva may be thickened as well.1
Melanocytic hamartomas of the iris called Lisch nodules are often seen (Fig. 58.7). They are smooth, dome-shaped lesions, typically yellow brown in color. Lisch nodules have been found in 92% of their affected population older than 6 years and in 100% of adults older than 21 years. They are specific to the diagnosis; however, their absence in young children does not rule out the disease.1
Glaucoma may occur at birth or early childhood, usually because of neurofibromatosis infiltration and obstruction of the drainage system. In addition, if the ipsilateral upper eyelid is affected with a plexiform neurofibroma, there is a 50% chance of glaucoma.1 Secondary angle closure glaucoma can also occur as a result of neurofibromatosis thickening of the ciliary body.14 The retina and optic nerve head may also be involved with hamartomas; in contradistinction to the uveal hamartomas, which are usually melanocytic nevi, the retinal and optic nerve head hamartomas are generally of glial elements.
Posterior to the papilla, the optic nerve may be involved by a glioma, which is different from the acquired gliomas of adulthood. The optic nerve glioma of neurofibromatosis is a true hamartoma, more accurately described as a juvenile pilocytic astrocytoma.23 They are generally benign, but malignant degeneration has been known to occur following radiotherapy.28 Gliomata of the anterior visual pathway (nerve and chiasm) may occur in 15% of patients with neurofibromatosis using CT scan criteria.29 In all patients with optic nerve gliomata, the incidence of neurofibromatosis is 25%. The glioma may produce proptosis and visual loss.30
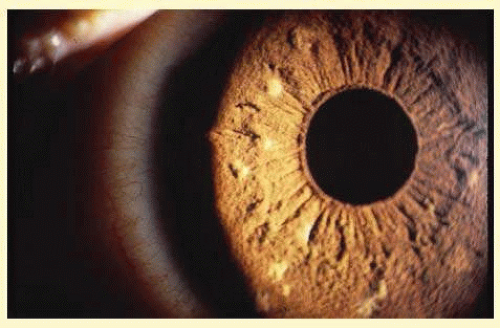 FIG. 58.7 Lisch nodules of the iris in a patient with neurofibromatosis. |
The hamartomas may also be found in the nonocular neural elements of the orbit, most commonly as plexiform neurofibroma or neurilemmoma, possibly causing proptosis (Figs. 58.4 and 58.8). Furthermore, absence of the greater wing of the sphenoid may occur, resulting in pulsatile exophthalmos.
Ocular involvement in NF2 is much less frequent and extensive than that in NF1. Between 60% and 80% of affected patients have cortical or posterior subcapsular opacities.14 Optic nerve meningiomas, epiretinal membranes, and retinal hamartomas associated with NF2 can cause vision loss as well.31 Intracranial tumors can also cause papilledema, leading to optic atrophy. Ocular motor disturbances can occur secondary to cranial neuropathies.1
Tuberous Sclerosis
This disorder is occasionally referred to as Bourneville disease, after Desiré Magloire Bourneville, the French physician, who described the triad of seizures, mental retardation, and cutaneous changes in 1880.32 The multiple “potatolike” tumors found in the brain gave rise to the term “tuberous sclerosis,” as named by Bourneville.
The reported incidence varies from 1 to 5 to 7 per 100,000.33,34 Similar to other autosomal dominant conditions, there is no gender predilection. About 40% of cases are inherited and 60% are sporadic.14 Penetrance is low and expressivity is variable; tumors have been reported in many organs and locations.35,36,37 Mutations are thought to be caused by two genes, TSC1 and TCS2, located at 9q34 and 16p13.3, respectively. Mutations of TSC2 are associated with more severe disease than those of TSC2.1
Systemic Manifestations
Systemic manifestations occur in the CNS, kidneys, and soft tissues.2 Seizures (motor, focal, petit mal, and psychomotor) occur in 80% to 90% of affected individuals. Autism, behavioral abnormalities, and mental retardation occur in 50% of all patients; virtually, all patients with mental retardation also have seizures.1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


