Ocular Developmental Anomalies
Scott E. Olitsky
Leonard B. Nelson
The clinical identification of an ocular developmental anomaly should stimulate curiosity concerning the etiology and pathogenesis of the condition. Etiology refers to the cause of the anomaly (e.g., a mutation, a teratogen, and so forth); pathogenesis refers to the mechanisms involved in the evolution of the anomaly. Because the mechanisms producing these anomalies often are related to a mishap in a particular stage of normal development, pathogenesis is best studied once normal ocular development is understood.
Mann1 divided the causes of developmental anomalies into four categories:
Category 1: This includes those anomalies attributed only to a mutation inherited by the fetus.
Category 2: This includes a genetic predisposition to malformations when exposed to certain environmental influences. Without this predisposition, the specific environmental influence alone would not produce maldevelopment. Conversely, without the genetic predisposition, the specific environmental factor would not be capable of inducing an abnormal morphology.
Category 3: This includes defects induced by environmental factors that interfere with the development of a genetically normal zygote. These factors may be infectious, chemical, or physical and are referred to as teratogens.
Category 4: This includes spontaneous mutations that result in ocular abnormalities that are not related to any known environmental agent.
Categories 2 and 3 indicate that genetic and environmental influences may interact to produce morphologic abnormalities. The same morphologic abnormalities may result from either the genetic or the environmental influence or from a variable mixture of both. When considering a structural abnormality, therefore, it may be impossible to determine accurately the proportionate contributions of the genetic and environmental factors that led to the production of the defect in question.
Because pathogenesis must be understood in the context of normal ocular development, a frame of reference is useful for discussion purposes. It is therefore customary to refer to “stages” of development. Of course, ocular development is a continuous process and does not proceed in discrete stages; however, staging remains a useful approach and will be used in this chapter. Three stages of fetal development will be addressed: embryogenesis, organogenesis, and differentiation.
In the initial stage, embryogenesis, the three germinal layers of the developing fetus—ectoderm, mesoderm, and endoderm—become organized. Defects during this period result in widespread somatic structural alterations, and the conceptus rarely proceeds to term. It is rare, therefore, for an event that occurs in embryogenesis to result in an isolated ocular anomaly. In organogenesis, the germinal layers throughout the fetus become organized into the general architectural patterns of the various organs. Therefore, many of the ocular anomalies arising from a mishap during this stage involve the entire globe (e.g., anophthalmia and microphthalmos with cyst). During the final period, differentiation, the characteristic substructures of each organ are developed; thus, abnormalities occurring during this period affect specific ocular structures. Specific structural ocular defects attributed to an event that occurs during this period may result from local growth retardation (e.g., microcornea), from the failure of embryologic structures to atrophy (e.g., persistent hyperplastic primary vitreous), or from an alteration in differentiation (e.g., retinal dysplasia).
There are two reasons to consult this chapter: (a) for an overview of ocular maldevelopment in the organized framework of embryology, and (b) as a reference for obtaining specific information about a particular entity. The remainder of this chapter is, therefore, divided into two parts. Part 1, The Relationship of Ocular Anomalies to Events in Development, is an outline of ocular developmental anomalies related to the periods of embryogenesis, organogenesis, and differentiation. Part 2, Developmental Ocular Anomalies, indexes and discusses ocular anomalies on the basis of the anatomic location of the defect. For rapid reference, a reader interested in a particular defect may turn directly to Part 2 for the discussion of an entity without first reading Part 1. The interested reader, however, should later return to Part 1 for a more complete grasp of the significance of the defect under study.
The Relationship of Ocular Anomalies to Events in Development
Stage 1: Embryogenesis
After fertilization, the ovum begins a rapid series of mitotic divisions, forming a solid ball of cells, the morula. These cells rearrange and organize around a central fluid-filled cavity, the blastocyst. The cells of the blastocyst divide, and those cells that will eventually produce the actual embryo accumulate at one pole, forming the embryoblast. These cells further differentiate into two layers, the epiblast and hypoblast. The two layers divide the central cavity of the blastocyst to form the amniotic cavity and the yolk sac. Central epiblast cells now invaginate between the two-layered embryoblast to form a tri-layered structure. The central cells of this structure differentiate to form the mesodermal layer, which fills in the area between the epiblast and hypoblast cells. The three definitive germ cell layers—ectoderm, mesoderm, and endoderm—have now been produced, and they will give rise to all other structures of the developing embryo. The period of embryogenesis, therefore, spans the events from fertilization through the organization of germinal layers.
Cephalad ectoderm differentiates into neural ectoderm, which then forms the neural plate; this, in turn, eventually develops into the head and brain. This neural ectoderm expands to form bilateral elevations called neural folds, which grow toward the midline and eventually fuse, producing the neural tube. Fusion begins in the central portion and continues anteriorly and posteriorly until only a small opening at either end remains. A malformation of the forebrain and mesodermal structures in the midline during the development of the neural plate may result in fusion of the structures that will eventually become the optic vesicles. Cyclopia refers to the condition in which complete fusion occurs of the elements of the two eyes (optic vesicles); partial fusion is called synophthalmia. The ocular anomalies produced during embryogenesis are merely reflections of protean systemic anomalies that are incompatible with life.
Stage 2: Organogenesis
The process of organogenesis begins with the segregation and arrangement of the primitive tissues to form the general pattern of the organs that will eventually develop. This process largely involves the movement of masses of tissues to the places where differentiation will occur.
During the third week of development, the optic pits or sulci appear as depressions or invaginations on the inner surface on each side of the anterior neural folds. As the neural folds approach each other, neural crest cells migrate from the neural ectoderm at its junction with the surface ectoderm. The neural crest cells migrate underneath the surface ectoderm and spread throughout the embryo, including the region of the optic pits. These cells are important precursors to many of the major structures of the eye, including corneal and iris stroma, ciliary muscle, choroid, sclera, and bone. Gradually, the primary optic vesicle forms as a spheric outpouching from the optic pit. The development of the optic vesicle from the optic sulcus occurs on approximately day. The optic vesicle is in contact with the surface ectoderm that will differentiate into the lens via the lens placode. Separation of the developing lens structure (lens vesicle) from the surface epithelium occurs at 33 days of gestation.
Primary anophthalmia results when the optic pit does not develop properly; it is not associated with a generalized fetal abnormality. One type of secondary anophthalmia involves an arrest in development after the appearance of the optic vesicle. Anophthalmia also may occur when the optic vesicle forms but subsequently degenerates. If the tip of the optic vesicle makes contact with the surface ectoderm over less than the normal area, a perfectly formed but microphthalmic eye results. On approximately day 28, the primary optic vesicle begins to invaginate to form the optic cup. If development proceeds to the outgrowth of an optical vesicle, but becomes arrested before its invagination, a condition known as congenital cystic eye will result.
Invagination of the optic vesicle occurs from the sides and below, simultaneously forming a groove along the underside of the optic cup and optic stalk; this groove is known as the fetal (choroidal) fissure. Immediately adjacent to the fetal fissure, tissue primarily of neural crest origin proliferates and forms the hyaloid system of vessels. The fetal fissure begins to fuse centrally, and this fusion extends both anteriorly and posteriorly. A typical coloboma occurs when the margins of the fissure fail to fuse anywhere along the line that extends from the optic disc to the inferonasal border of the pupillary margin. Another defect that results from an anomalous closure of the fetal fissure is microphthalmos with cyst formation. Optic nerve aplasia also has been related to anomalous closure of the ventral fissure.
Stage 3: Differentiation
The events of organogenesis result in the framework of the developing eye. The specific components of the eye are developed by the process of differentiation.
Lens Differentiation
Between days 24 and 26, the surface ectoderm adjacent to the optic vesicle thickens, forming the lens plate (placode). The lens placode invaginates to form the lens vesicle, which has a hollow center, between days 32 and 33 (Fig. 53.1). The lens vesicle consists of a single layer of cells whose apices are directed toward the center. After the lens vesicle has detached from the surface ectoderm, it is surrounded by a basal lamina, which will become the lens capsule.
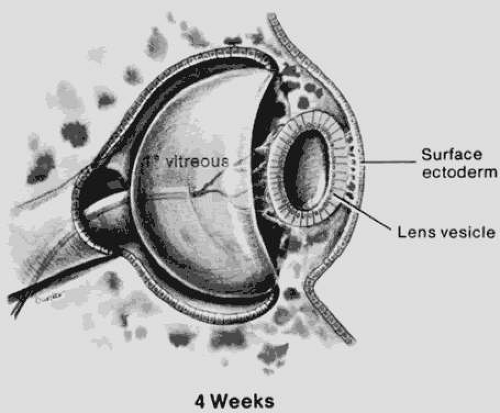 Figure 53.1. The eye at 4 weeks. The lens vesicle is about to separate from surface ectoderm. The optic vesicle is an invaginated cup. Note the relationship of the vascularized primary vitreous to the lens. (Courtesy of Irene H. Maumenee, MD.) |
Primary aphakia results when the lens plate is absent.2 A lens that forms but subsequently degenerates and largely disappears leads to secondary aphakia.2 If the lens plate forms normally, cuboidal cells line the anterior part of the lens vesicle. The posterior portion of the vesicle is lined by columnar cells that form the primary fibers. These fibers fill the lens vesicle, obliterating its cavity and creating an arc across the lens anterior to the equator. The anterior lens epithelial cells remain cuboidal and become the permanent lens epithelium that, through mitosis, gives rise to future cortical lens fibers later in life.
By the seventh week of development, secondary fibers begin to proliferate in both directions from the equatorial region of the lens. The secondary fibers interlace anteriorly to produce an upright Y suture and posteriorly to produce an inverted Y suture. The lens continues to grow throughout a person’s life through the proliferation of secondary lens fibers. As the fibers are laid down, the different optical densities of the fibers produce four zones of discontinuity that can be observed clinically with a slit lamp:
The embryonic nucleus, an optically clear central area formed in embryonic life (during months 1 to 3) from the primary lens fibers, which retain embryonic transparency
The fetal nucleus, formed from secondary fibers from the third to eighth month of fetal life
The infantile nucleus, formed from the last weeks of fetal life through puberty
The adult nucleus, formed after puberty in adult life
Abnormalities of the lens that occur during the stage of differentiation include alterations in size (e.g., microspherophakia), in shape (e.g., anterior and posterior lenticonus), and in the lens fibers themselves (e.g., developmental cataracts).
The developing lens receives its blood supply from multiple branches of the hyaloid artery. These cover the anterior and posterior aspect of the lens and anastomose with vessels within the mesenchyme tissue that covers the lens, forming the pupillary membrane. This hyaloidal vascular system is called the tunica vasculosa lentis. Its greatest development occurs at 9 weeks’ gestation, starts to regress by the end of the third month, and is usually complete by the eighth month. Failure of this system to regress leads to a persistent pupillary membrane. A Mittendorf dot is also a failure of complete regression and represents that part of the hyaloid artery that attached to the posterior lens capsule.
Retinal Differentiation
When the optic vesicle collapses, the neural ectoderm folds onto itself to produce the optic cup, which has a double layer of neural ectoderm. The optic cup will differentiate into the neurosensory retina (inner layer of neural ectoderm) and retinal pigment epithelium (outer layer). Further development of the sensory retina may be divided into four stages of progressive differentiation: the initial differentiation into zones, the organization into temporary layers, the migration and differentiation of cells, and the final organization of layers. Disorganized differentiation may result in the histologic picture of retinal dysplasia.3
Vitreous Differentiation
The primary vitreous forms between the inner layer of the optic cup and the developing lens at approximately 6 weeks. The secondary vitreous is derived from the retina and surrounds the primary vitreous. It is composed of very fine, densely packed fibrils that are arranged in an orderly manner at right angles to the retinal surface. Condensation of the fibrils forms the walls of the canal of Cloquet, which is a conduit for the hyaloid artery. The hyaloid artery supplies the vascular network surrounding the lens. At the end of the third month, the hyaloid artery atrophies and retracts with the primary vitreous to form a narrow and constricted zone in the central vitreous cavity (Figs. 53.2 and 53.3). The atrophy begins centrally and proceeds anteriorly and posteriorly. Incomplete atrophy may result in anterior hyaloid remnants associated with the lens or in posterior remnants associated with the optic nerve head. An example of an anterior remnant is the Mittendorf dot. Bergmeister papilla may be considered a posterior remnant of the hyaloid system.
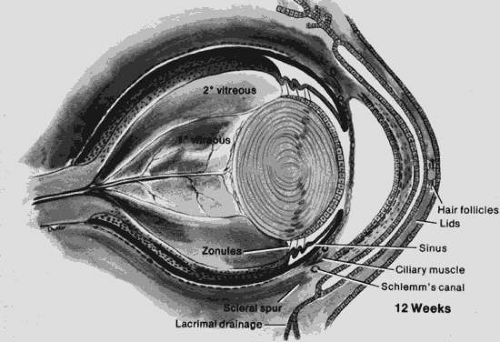 Figure 53.2. The eye at 12 weeks. The cornea is separated from the more anterior fused eyelids. A membrane covers the pupil (pupillary membrane). Note the relationship of the scleral spur to the anterior chamber; the angle is incompletely formed. The lens vesicle is now obliterated. The secondary vitreous is avascular, whereas the primary vitreous is vascularized. Note the vascular arcades on the posterior surface of the lens (posterior tunica vasculosa lentis). (Courtesy of Irene H. Maumenee, MD.) |
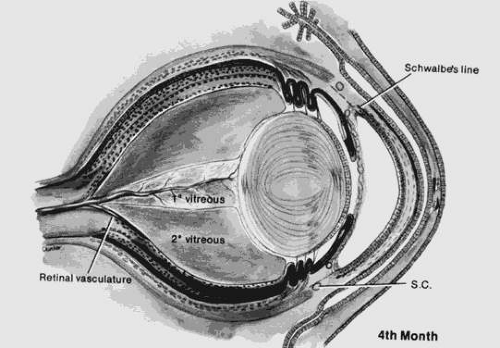 Figure 53.3. The eye at 4 months. Note the changed proportions of secondary and primary vitreous. (Courtesy of Irene H. Maumenee, MD.) |
The third period of vitreous formation begins at the end of the third month. The tertiary vitreous begins as an accumulation of collagen fibers between the equator of the lens and the optic cup; it will eventually differentiate into the vitreous base and lens zonules.
Scleral Differentiation
The sclera is derived from tissue of neural crest origin. Most of the cornea is derived from the same mesenchymal tissue with the exception of the corneal epithelium, which is derived from surface ectoderm. The anterior sclera forms first as a condensation of this tissue and is continuous with the cornea. Condensation proceeds posteriorly until the twelfth week of development, at which time the sclera surrounds the optic nerve.
Corneal Differentiation
The lens vesicle detaches from the overlying surface ectoderm during the sixth week of gestation. During the seventh week, a single layer of neural crest cells invades the region between the surface ectoderm and the lens vesicle. This single layer of cells will differentiate into the corneal endothelium. A second wave of neural crest cells migrates between the primitive endothelium and surface ectoderm to form fibroblasts. These fibroblasts will eventually differentiate into keratoblasts that will produce the collagen fibrils and extracellular matrix of the corneal stroma.4 Initially, these neural crest cells occupy the space that will become the anterior chamber. They are the precursor cells that will develop into the corneal stroma, corneal endothelium, anterior iris stroma, and many of the structures of the iridocorneal angle. The surface ectoderm will give rise to the corneal epithelium. The anterior chamber is formed when these neural crest cells separate from the tunica vasculosa lentis and pupillary membrane, which overlie the anterior lens surface. Therefore, the anterior chamber is derived almost entirely from neural crest cells, not mesoderm, as was once believed. The vascular endothelium is now thought to be the only component of the anterior segment that is derived from mesoderm.
The corneal epithelium is derived from surface ectoderm, whereas the remainder of the cornea is derived from neural crest cells, as described previously. The corneal stroma gives rise to the Bowman membrane. Therefore, the Bowman membrane is of neural crest origin, and is a condensation of anterior corneal stroma. It is not the basement membrane of the corneal epithelium and is not derived from the same precursor cells. The Descemet membrane is present by the sixth month of gestation, and the cornea begins to become transparent at about this time.
Iris and Ciliary Body Differentiation
During the third month of development, the anterior rim of the optic cup, which is composed of neural ectoderm, begins growing rapidly in a curve around the anterior surface of the lens. The iris pigmented and nonpigmented epithelium will develop from this anterior advancing edge of the optic cup. The iris sphincter and dilator muscles develop from these epithelial layers and are the only muscles of the body that are derived from neural ectoderm. The inner epithelial layer begins to develop folds, which are the early ciliary processes. The ciliary body epithelium also develops from the anterior portion of the optic cup. The tertiary vitreous forms from an extracellular matrix that is produced by the ciliary epithelium and will eventually become the lens zonules. The iris stroma develops from the neural crest cells, which migrate between the lens vesicle and surface ectoderm.
Iridocorneal Angle Differentiation
Traditionally, the study of the development of the anterior chamber and angle has concentrated on the role of mesenchyme. It is now known that neural crest cells are the major contributor to the development of these structures. There continues to be some controversy, however, regarding the mechanism by which the angle is formed. There are conflicting opinions as to whether the angle structures are formed as a result of simple cleavage of the iris root from the cornea, or whether a more active growth process is required for normal angle development.
Optic Nerve Differentiation
The optic nerve develops within the substance of the optic stalk. The nerve fibers of the developing optic nerve are composed of axons of the ganglion cells in the retina. The nerve fibers completely fill the optic stalk by the end of the seventh week. Myelination of the nerve fibers starts at the chiasm at about 7 months and progresses anteriorly toward the eye. Myelination normally stops at the lamina cribrosa approximately 1 month after birth. Myelinated nerve fibers occur if the myelination process continues past the lamina and most likely represents ectopic myelination, not a structural defect in the lamina cribrosa.
Eyelid and Lacrimal Apparatus Differentiation
The eyelids develop from both surface ectoderm and neural crest cells. Surface ectoderm gives rise to the epidermis, cilia, and conjunctival epithelium, whereas the deeper structures of the lids, including the dermis and tarsus, are derived from neural crest cells. The orbicularis and levator muscles develop from mesoderm. The eyelids begin to form by 6 weeks and grow to cover the eye. The upper and lower folds fuse at approximately 12 weeks and gradually start to separate at 6 months’ gestation.
The lacrimal apparatus is formed by contributions from the lateral nasal and maxillary processes as well as from the lids. During the third month, the lacrimal canaliculus is formed by the lysis of a central core of cells. The lacrimal punctum opens onto the lid margin just before the lids separate, usually during the seventh month. The lower end of the nasolacrimal duct frequently is separated from the cavity of the inferior meatus at birth by a membrane. This membrane consists of the opposed mucosal linings of the nasal fossa and the lower end of the duct. Obstruction of the nasolacrimal duct usually is caused by defective canalization or persistence of the nasal mucosal membrane.5
Developmental Ocular Anomalies
Anophthalmia
Anophthalmia (Fig. 53.4) is the congenital absence of the eye. It is a rare entity and occurs in approximately 2 in 100,000 births.6,7 Anophthalmia can be divided into three types; (a) primary, which results from an isolated failure of optic pit development (i.e., the remainder of the neural tube may be unaffected); (b) secondary, which results from a complete suppression or abnormality of the forebrain, in which case the anophthalmia is merely a component of neural malformation; and (c) consecutive, or degenerative, which results from regression of a formed but rudimentary optic vesicle.
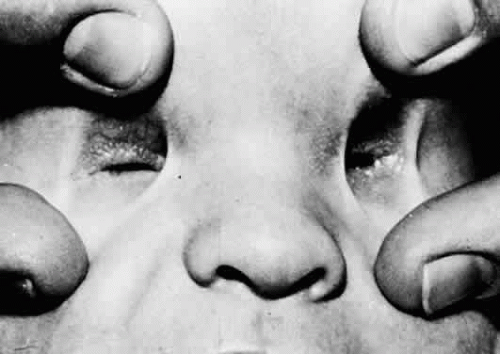 Figure 53.4. Bilateral clinical anophthalmia. No eye is apparent in either orbit. |
Because anophthalmia may be difficult to distinguish clinically from profound microphthalmos, removal of the orbital contents with serial histologic examination may be necessary to establish the diagnosis. A small congenital cystic eye (see later discussion) also may give the clinical appearance of anophthalmia. Because few cases are confirmed histologically, the term clinical anophthalmia is used to describe the condition in which an eye appears to be absent. Radiologic and ultrasonographic examination may be helpful in attempting to distinguish anophthalmia from severe microphthalmos or when searching for a small cystic eye within the orbit. Systemic malformation syndromes occur in many patients with clinical anophthalmia, and unilateral cases often show abnormalities of the contralateral eye.8,9
Most cases of anophthalmia occur sporadically, although autosomal-dominant, autosomal-recessive, and sex-linked–recessive transmissions have been reported. The causes of developmental failure of the optic vesicle or vesicle regression may include environmental factors, such as radiation or anoxia. Animal models of anophthalmia suggest that suppression of the optic vesicles may result from the introduction of a variety of teratogenic factors, such as lithium, trypan blue, or actinomycin, at the time the optic vesicle is beginning to appear.
Cyclopia and Synophthalmia
Cyclopia (total fusion of the optic vesicles) and synophthalmia (partial fusion of the optic vesicles) represent degrees of an anomaly that prohibits the development of separate eyes. They occur in approximately 1 in 100,000 births.10 Two mechanisms can account for either of these developmental malformations: an early defect, in which a single bilobed midline region fails to separate completely; or a later loss of the midline territory, which leads to fusion of the ocular fields that had been previously divided. Because the prosencephalon is responsible for the development of separate eyes, cyclopia and synophthalmia are anomalies that reflect profound neural maldevelopments that are incompatible with life.
The cyclopic eye usually demonstrates multiple internal abnormalities, such as the presence of retinal rosettes, the absence of the retinal pigment epithelium (RPE) at the posterior pole, and abnormal choroidal development.11 Synophthalmia is characterized by graded degrees of ocular fusion. Except for the anterior structures of the eye, such as the lens and anterior uvea, the fused eyes may share a single structure (e.g., a common optic nerve).12,13 In most cases, the retina may show a coloboma of various degrees, rosette formation, and a paucity of ganglion cells.
Teratogenetic agents and chromosomal abnormalities both may cause cyclopia and synophthalmia. The teratogenetic periods in the human are probably at 2.5 weeks’ gestation for cyclopia and at 3 weeks’ gestation for synophthalmia.14 A deletion on the long arm of chromosome 7 has been implicated as an etiologic factor in arresting prosencephalon development at an early stage, leading to the development of cyclopia.15 Cyclopia has been reported in association with trisomy 4.16
Congenital Cystic Eye
Congenital cystic eye results when the optic vesicle fails to invaginate. When invagination does not occur, the development of the entire globe is arrested, and the ectodermal elements (what will become the cornea and lens placode) do not differentiate. Instead of a globe, the orbit contains a cyst, which may be either unicameral or subdivided into several loculi.17 If the congenital cystic eye is very small, it may be confused with clinical anophthalmia (see earlier discussion). Congenital cystic eyes also may be confused clinically with the condition of microphthalmos with cyst, in which the cyst is so large that it dwarfs the severely microphthalmic component.
Microphthalmos
Microphthalmos is a general term used to describe an eye that is smaller than normal. It can result from an insult that may occur at various developmental stages. An early insult may lead to a primary decrease in the size of the optic vesicle. Because the growth of the sclera and choroid appears to depend on intraocular pressure (IOP), insults that occur later in development that affect proper maintenance of the IOP may lead to a secondary decrease in the size of the fetal eye.18 An example of this can be seen in colobomatous microphthalmos, in which failure or late closure of the embryonic fissure prevents the establishment of normal fetal IOP (see later discussion). Microphthalmos occurs in many forms and can be divided into three basic types: (a) pure, (b) simple, and (c) complex.
Pure Microphthalmos (Nanophthalmos)
Pure microphthalmos, also called nanophthalmos, refers to a small eye containing a lens of “normal” volume. It is thought to result from an arrest in ocular development after the fetal fissure has closed. The eye is small in its overall dimensions, but shows no other gross defects. Because the size of the lens is normal, patients with nanophthalmic eyes have a shallow anterior chamber and pronounced hypermetropia (more than 10 diopters). Therefore, these eyes are prone to attacks of angle-closure glaucoma.19 Thickening of the sclera around the exits of the vortex veins may contribute to retinal and choroidal effusion.20,21 Scleral thickening is a result of the abnormal interlacing of the scleral fibers, which may be secondary to abnormal glycosaminoglycan metabolism.22,23 Histopathologic evaluation of the sclera in nanophthalmos has demonstrated abnormal collagen fibrils that were frayed, split, and contained lightly stained cores.24 In early life, the visual acuity is usually normal, although macular hypoplasia may account for poor vision in some younger patients. Poor vision later in life is often due to the secondary complications of this disorder. Nanophthalmos is generally bilateral, and these eyes are usually deeply set in shallow orbits.
Most affected individuals are members of pedigrees that demonstrate consanguinity. The mode of inheritance, therefore, is presumed to be autosomal recessive. Only a few pedigrees compatible with autosomal-dominant inheritance have been reported.25
Simple Microphthalmos
Simple microphthalmos is the term used to describe a small eye that is otherwise normal. Approximately 50% of patients with simple microphthalmos have an associated systemic developmental abnormality. These eyes have a short axial length and are therefore moderately hyperopic. The level of hyperopia is less than that seen in nanophthalmos (usually less than +10.00 diopters).26 Most patients have normal vision. The complications that are seen with nanophthalmos do not occur in this disorder.
Most cases of simple microphthalmos are sporadic and occur bilaterally. Simple microphthalmos represents a retardation in growth that occurs once the primary optic vesicle has formed and invaginated. It has been caused in animal experiments by radiation, mechanical stimulation, and chemicals. It also may be associated with systemic disorders, such as fetal alcohol syndrome, myotonic dystrophy, and achondroplasia.
Complex Microphthalmos
The term complex microphthalmos denotes a small, deformed eye. One or more malformations may occur as an isolated ocular entity or as part of an associated systemic disorder. Associated ocular deformities can include anterior segment maldevelopment, hyperplastic primary vitreous, and other abnormalities of the lens, vitreous, or retina. Complex microphthalmos may be unilateral or bilateral, and the vision can range from normal to no light perception, depending on the severity of the associated defects. Both sporadic and hereditary forms are known.
Microphthalmos with coloboma is one specific type of complex microphthalmos and is caused by an incomplete closure of the embryonic fissure. It also may occur as both a sporadic and hereditary form. It may be associated with a number of systemic diseases, including the coloboma, heart defect, atresia cochonal, retarded growth, genital hypoplasia, ear anomalies (CHARGE) syndrome. Recognition of this entity when evaluating a child with microphthalmos is important, because the heart defects in the CHARGE association can be lethal. Autosomal-dominant colobomatous microphthalmos is a well-known and important disorder. It occurs without an associated systemic abnormality and has variable expressivity and penetrance. Family members of affected patients should receive appropriate genetic counseling.
Microphthalmos with cyst results from the proliferation of neuroectoderm at the edge of a persistent embryonic fissure. The cavity of the cyst is continuous with the interior of the microphthalmic globe. Rudimentary optic nerve fibers can be seen within the cyst wall.27 The microphthalmic eye itself often is overshadowed by the larger orbital cyst (Fig. 53.5).28 Cases of extreme microphthalmos may be confused with anophthalmia. The anterior segment of the microphthalmic eye may appear normal, although it commonly displays marked disorganization and iridocorneal adhesions. The lens is frequently cataractous and dislocated.29 The retina usually is detached, disorganized, and gliotic.30 No consistent association has been documented between microphthalmos with cyst and a specific systemic anomaly. Systemic disorders reported with microphthalmos with cyst include central nervous system (CNS) defects (e.g., meningoencephalocele, hydrocephalus) and cardiac, urogenital, facial, and skeletal abnormalities.31
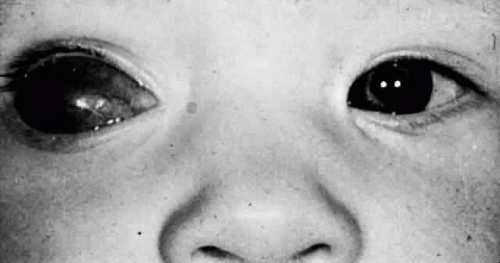 Figure 53.5. Microphthalmos with cyst. (Courtesy of William H. White, MD.) |
Typical Colobomas (of the Iris, Retina, Choroid, and Optic Nerve)
The term coloboma is employed in ophthalmology to refer to any notch, gap, or fissure in any of the ocular structures, whether it be congenital or acquired. Technically, therefore, a surgical iridectomy is a type of iris coloboma, but for the remainder of this discussion, the term coloboma will refer only to congenital defects.
Colobomas may occur anywhere along the embryonic fissure and can therefore affect the iris, ciliary body, choroid, retina, and optic nerve. The embryonic fissure begins to close at the equator of the eye sometime around the fifth week of gestation. Closure of the fissure then proceeds in an anterior and posterior direction. Because of this, colobomas may be found at the two ends of the embryonic fissure—the iris and optic nerve—with normal tissue between them.
Iris colobomas can be designated as typical or atypical, depending on where they are located. Typical iris colobomas are located inferonasal to the pupillary border; thus, they reflect the incomplete closure of the embryonic fissure. Iris colobomas may be associated with flattening of the exposed portion of the lens, gaps in the zonular fibers, and localized conical lens opacities. The lens notch is commonly referred to as a lens coloboma. This is not a true coloboma because no focal absence of lens material is present, and the lens does not develop from the embryonic fissure. Rather, it results from the absence of zonules and zonular traction in the region of the ciliary body coloboma. Atypical iris colobomas are not located in the inferonasal sector, and they are not considered to be related to defective embryonic fissure closure. Atypical iris colobomas are therefore not associated with posterior uveal or optic nerve colobomas.
A chorioretinal coloboma appears through the ophthalmoscope as a glistening white defect with distinct margins, often outlined by irregular pigment clumps. Although a coloboma may occasionally be flat and smooth, the floor of the defect is usually uneven and usually bulges posteriorly. An intercalary membrane of undifferentiated neurosensory retina may cover the defect. Breaks in this membrane have been reported to have caused retinal detachment. When a hemorrhage is found in the coloboma, a break in the intercalary membrane may be present.32
In optic nerve coloboma (Fig. 53.6) the optic disc is enlarged, frequently oval vertically, and excavated. The colobomatous defect may involve the entire disc or just the inferior portion. The excavated region is decentered interiorly and may extend to involve the choroid and retina. When it does, the eye is often microphthalmic.33 When the entire disc is involved, the inferior region is excavated to a greater extent than is the remainder of the disc, confirming its colobomatous nature. The retinal vessels may radiate from the disc in a spoke-like fashion, with fewer bifurcations than normal. An association between optic nerve colobomas and basal encephaloceles has been reported34; however, examination of the literature reveals only a few photographically documented cases, and the reported association may be because of its confusion with morning glory disc anomaly.
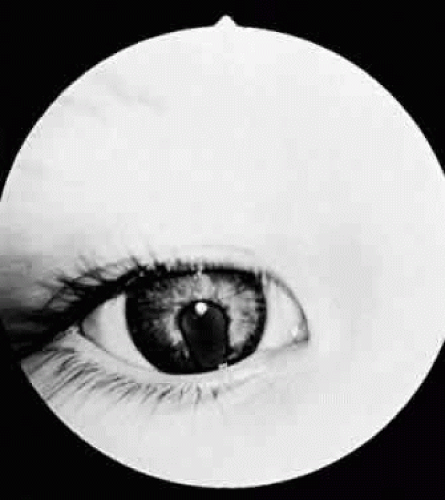 Figure 53.6. Optic nerve coloboma. |
Vision in the presence of a colobomatous defect can range from normal to no light perception. The degree of vision loss is related to the location of the defect and the presence of any associated abnormalities. Isolated iris colobomas do not usually affect visual acuity, whereas optic nerve colobomas almost always lead to some reduction in vision. If a chorioretinal coloboma is located in the posterior pole, an associated staphyloma may cause disturbance of the macula with resultant poor vision.
Isolated colobomatous anomalies are inherited most commonly in an autosomal-dominant manner with highly variable penetrance and expressivity.35 Because colobomas may be associated with a variety of systemic anomalies and heritable disorders (Table 53.1), affected individuals should be examined systemically and a careful family history should be taken. A thorough genetic evaluation should be performed when indicated.
TABLE 53.1. Coloboma and associated disorders. | ||
|---|---|---|
|
Lens Anomalies
Congenital Aphakia
Congenital aphakia, a very rare anomaly, can be divided into two types: primary, in which a lens failed to develop; and secondary, in which a lens developed to some degree, but either was resorbed or extruded through a corneal perforation before or during birth.2 Primary congenital aphakia has been characterized histologically by aplasia of the anterior segment. This defect may result from anomalous invagination of the optic vesicle, which disturbs the normal relationship between the optic vesicle and the surface ectoderm. The lack of surface ectodermal induction may lead to widespread anterior and posterior segment anomalies.36 The abnormal optic vesicle invagination also may be accompanied by defects in the closure of the fetal fissure, and therefore by typical colobomas.
Secondary congenital aphakia is more common than is the primary type. The lens is usually represented by a wrinkled capsule. Several attempts have been made to explain secondary aphakia. One theory suggests that the lens may develop normally, but that the capsule is thinner than usual and ruptures. Intrauterine inflammations also may lead to degeneration of the lens and to subsequent capsular shrinkage, which is sometimes accompanied by mesenchymal infiltration of the lens remnant.
Microspherophakia
Microspherophakia is a bilateral condition in which the lens is usually small and spheric. Following pupillary dilation, the lens edge may be identified with a slit lamp. The small size and spheric shape of the lens may result in significant myopia. The elongated zonules may permit the lens to move forward, increasing the area of iridolenticular contact.37 This mechanism may lead to pupillary block. Miotics may increase the pupillary block, and mydriatics may relieve it, a paradox that Urbanek38 termed inverse glaucoma and that others have confirmed exists.39,40 Microspherophakia has been reported in association with such systemic entities as Marfan syndrome, homocystinuria, Alport syndrome, Klinefelter syndrome, and Weill-Marchesani syndrome.41,42,43,44 It may occur as an isolated anomaly transmitted as either an autosomal-dominant or autosomal-recessive condition.45 The lens is typically spheric before the fifth or sixth month of fetal life, so an arrest in lenticular development at this stage would explain uncomplicated microspherophakia.
Anterior Lenticonus
Anterior lenticonus is a rare bilateral condition in which the anterior surface of the lens bulges centrally. It may be accompanied by lens opacities or other eye anomalies and is a prominent feature of Alport syndrome.46,47 Examination by retinoscopy or by ophthalmoscopy shows a dark area resembling an oil droplet in the center of the lens. The increased curvature of the central area may cause high myopia.
Histologic examination of the anterior capsule in anterior lenticonus shows it to be one-third the normal thickness, having multiple capsular dehiscences containing fibrillar material and vacuoles.48,49 The capsular fragility observed in this condition is a result of a mutation in the gene that encodes for one or more chains in type IV collagen.50,51,52 The fragility of the anterior capsule most likely forms the basis for the progressive lenticonus and anterior polar cataract, as well as the susceptibility of the lens capsule to rupture, either spontaneously or after minor trauma.53
Posterior Lenticonus
Posterior lenticonus, which occurs more commonly than does anterior lenticonus, is an ectasia of the posterocentral surface of the lens. It often is associated with progressive lens opacities of the posterior cortex. The majority of cases are unilateral, and the condition is found more frequently in females than in males. When no lens opacities are present, the clinical appearance of posterior lenticonus resembles the oil droplet appearance of anterior lenticonus. The characteristic localized posterior bulge can be seen with a slit lamp. The etiology of posterior lenticonus is unknown. Because the zonules exert traction on the lens and determine its shape, focal defective zonular traction along the posterior lens capsule may account for abnormal curvature in the area.
Developmental Cataracts
Developmental cataracts may be classified according to the location (e.g., nuclear, polar) or configuration (e.g., coralliform, blue dot) of the opacity. Progress has been made in determining the etiology of many congenital cataracts. The various factors that might cause these lens defects include hereditary factors, infectious agents, metabolic abnormalities, toxins, endocrinologic effects, and ionizing radiation. Despite a meticulous medical history and a thorough clinical and laboratory workup, the cause of a congenital cataract in any particular patient can still escape identification. Because they may develop at any time during the life of the fetus, lens opacities often make excellent markers for the timing of developmental defects. In general, developmental aberrations affecting the primary lens fibers may cause central lens opacities. If the secondary fibers are opacified, a clinical opacity restricted to a zone (zonular cataract) may occur.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


