Albinism
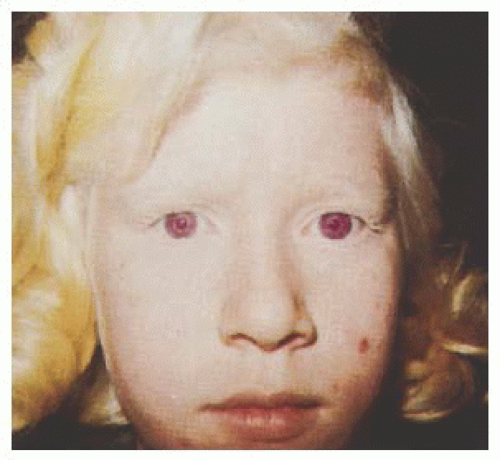
Albinism represents a group of inherited disorders characterized by a congenital reduction of melanin pigment in the developing eye. Specific changes in the eye and visual system result from this pigmentary defect and are common to all types of albinism. Although the reduction in melanin synthesis can be localized to the eye (ocular albinism [OA]), it is much more likely to involve the skin, hair, and eye (oculocutaneous albinism [OCA]) (
Fig. 21.1). Affected individuals show hypopigmentation of skin and hair with characteristic eye involvement. Therefore, the diagnosis is usually made on the basis of the clinical findings (
1).
Melanin is exclusively produced by a relatively small population of melanocytes with two embryonic origins. Melanocytes deriving from the neural crest migrate to and settle in the skin, hair, and eye (choroid and iris). Melanocytes in the retinal pigment epithelium (RPE) originate from the outer layer of neuroectoderm that makes up the optic vesicle. Production of melanin occurs in specialized intracytoplasmic organelles known as melanosomes. These membrane-bound organelles contain the enzymes needed to convert tyrosine to melanin. Hydroxylation of tyrosine to dihydroxyphenylalanine is mediated by tyrosinase. Then additional enzymes, including tyrosinase-related proteins 1 and 2, regulate subsequent oxidative steps in the pathway, resulting in the synthesis of eumelanin and pheomelanin, the two major forms of melanin (
1).
Proteins other than the family of tyrosine enzymes are involved in melanogenesis. The OCA2 locus in humans (pink eye dilution gene in mice) encodes for a transmembrane protein important for the synthesis of melanin (
2). Mutations within this locus are associated with OCA2 and a subset of patients with Prader-Willi syndrome (PWS) and Angelman syndrome (AS). This locus has been mapped to chromosome l5q. The genes associated with Chédiak-Higashi (
CHS1) and Hermansky-Pudlak (
HPS1-8) syndromes encode for proteins involved in the biogenesis and trafficking of melanosomes and other lysosomal-related organelles. Combined abnormalities of melanosomes, along with lysosomal and other lysosomal-related organelles (platelet-dense granules, basophil granules, major histocompatibility complex class 2 compartments), in these genetic disorders demonstrate the shared properties of these organelles (
3).
Congenital nystagmus with an abrupt onset during the first 3 months of life is usually the presenting clinical sign. The nystagmus has a pendular or jerk waveform or can evolve from a pendular to jerk waveform. Nystagmus severity can be invariant or vary with horizontal gaze position. Patients with gaze position differences will often adopt a compensatory head turn to align the target at this eccentric gaze position where retinal slip is minimized and visual acuity is optimized. Unstable fixation and immature tracking in infancy can lead to vision concerns. Despite delays in acuity development, final visual acuities range from 20/40 to 20/200.
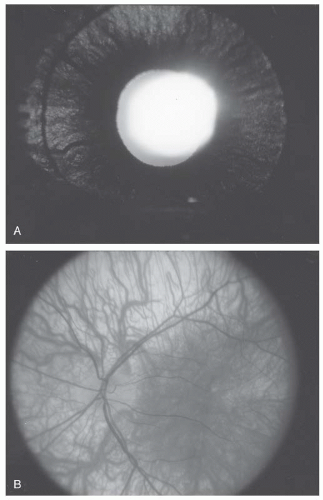
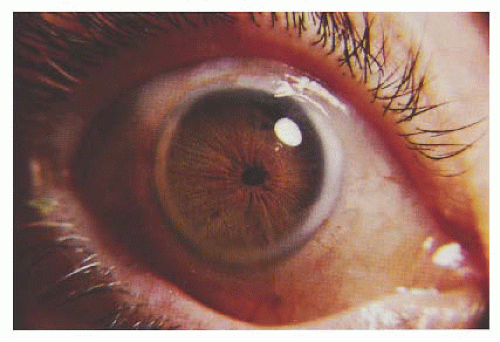
Iris hypopigmentation is an important diagnostic finding observed in most but not all albinism patients. Incident light reflected from within the eye can penetrate the iris in albinism, owing to reduced melanin in its posterior pigmented layer. Diffuse iris defects can be grossly detected by transscleral illumination using a light source placed on the bulbar conjunctiva, but punctate defects are subtle and best appreciated at slit-lamp examination (
4) (
Fig. 21.2A).
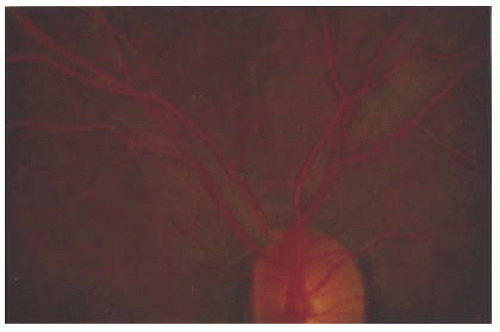
Fundus pigmentation is typically commensurate with skin pigmentation; in albinism, the fundus is hypopigmented. Loss of melanin pigmentation within the RPE allows for direct visualization of the underlying and prominent choroidal vessels (
Fig. 21.2). The macula, unlike the peripheral retina, contains luteal (carotenoid) pigments. Since the luteal pigments are intact in albinism, the hypopigmentation is more conspicuous in the peripheral retina than the macula. Functionally, the RPE is intact, and the full-field electroretinogram (ERG) is normal.
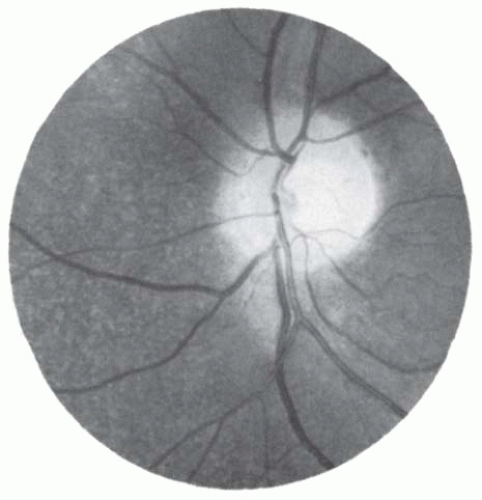
The hallmark of albinism is macular hypoplasia (
Fig. 21.2B). The normal eye is characterized ophthalmoscopically by the presence of a concave depression in the region of the macula, owing to the lateral displacement of the inner retinal layers. In albinism, the macula is coplanar with the surrounding retina, causing loss of the macular and foveal light reflexes, and blood vessels may course through this normally avascular structure. As a result of the loss of ganglion cells originating from the fovea, the optic disk contains fewer axons and its diameter tends to be smaller but within the normal range. The anatomic abnormalities of the macula especially the blunted macula reflex, due to abnormal persistence of the inner retinal layers, and reduced density of cone photoreceptors are best characterized with optical coherence tomography (OCT) (
5).
The visual pathways are abnormal in albinism. Melanin plays an important developmental role in the routing of optic nerve axons. Normally, the ratio of fibers that project to the contralateral hemisphere versus those that project to the ipsilateral hemisphere is 53:47. In albinism, the ratio is increased because axons from the temporal retina are routed contralaterally rather than ipsilaterally. This leads to altered binocular representation of the visual scene in which spatial correspondence in the retina is not maintained in the visual cortex. If the two retinal images are not in precise cortical registration, then binocular interactions cannot develop normally, and stereopsis is severely reduced or absent. Because stereopsis provides a feedback signal that helps to establish and maintain eye alignment, albinos frequently have strabismus. Misrouting of optic axons can be demonstrated in albinism using a lateralizing visually evoked potential (VEP) (
4). The response to a pattern-onset stimulus presented monocularly is recorded with electrodes offset to the right and left of the midline. Asymmetric amplitudes between the two hemispheres are indirect evidence of abnormal decussations. Because the various types of albinism share overlapping clinical features, albinism is most reliably classified by the underlying molecular defect (
Table 21.1). In OCA1A, there is a complete lack of tyrosinase activity. Individuals with OCA1A are usually diagnosed at birth on the basis of having white scalp hair, white skin, and blue irides, especially in dark-complexioned families. Nystagmus, reduced acuity, and strabismus may be the initial manifestations in light-complexioned families in whom pigmentary differences are less conspicuous. The skin and hair remain white throughout life, visual acuity ranges from 20/100 to 20/400, and no melanin pigmentation develops within the iris or retina.
In OCA1B, tyrosinase activity is reduced or temperature dependent (
6). Although affected individuals have white or light yellow hair and white skin at birth, the skin, hair, and eyes acquire pigmentation by the age of 1 to 3 years. At slit-lamp examination, peripapillary clumps or radial spokes of pigmentation become evident, along with fine granular pigmentation of the retina. Despite these pigmentary changes, there is typically no improvement in visual acuity.
Individuals with OCA2 have a defective P protein with normal tyrosinase activity (
2). The
OCA2 gene encodes for the P protein, which is an integral component of the melanosomal membrane. These individuals produce some melanin but predominantly yellow pheomelanin rather than black-brown eumelanin. The phenotype is determined by the relative amounts of pigmentation of skin, hair, and eyes, which can range from minimal to near normal. In general, the pigmentary deficiency is less severe than that observed in OCA1A but can overlap with that in OCA1B. Scalp hair and skin coloration vary from off-white to blond to brown. The ocular findings are identical except for the increased amounts of iris and retinal pigmentation. Visual acuity ranges from 20/30 to 20/400 but is usually near the 20/200 level (
1).
PWS and AS have hemizygous deletions within chromosomal locus 15q where the
OCA2 gene colocalizes (
1,
7). Individuals with PWS and AS can have hypopigmented skin and hair, but the ocular features of albinism are usually lacking. When the ocular findings of albinism are found, affected individuals are reported to have an
OCA2 gene mutation of the nondeleted chromosome (
8). PWS is characterized by hypotonia, obesity, mental retardation, hypogonadism, short stature, and small hands and feet. The diagnosis of AS should be suspected in any albino with microcephaly, developmental delay, inappropriate laughter, and seizures. Craniofacial stigmata include flat occiput, thin upper lip, prominent jaw, and widely spaced teeth.
OA has an X-linked inheritance pattern and is less prevalent than OCA. Affected males have normal skin and hair pigment along with ocular features of albinism. Despite normal skin appearance, light and electron microscopy demonstrate aggregates of abnormal melanosomes within keratinocytes and melanocytes in affected males and carrier females. The ocular findings include congenital nystagmus, reduced visual acuity (20/40 to 20/200), hypopigmentation of the iris and retina, and foveal hypoplasia. In individuals with dark complexions, the iris and retinal pigmentary changes can be subtle or absent, and there is less severity of foveal hypoplasia and reductions in visual acuity. The
OA1 gene encodes a G-protein-coupled membrane receptor that localizes to melanosome membranes (
9). The obligate ligand for the putative OA1 receptor has not been identified but is likely important to melanosome function.
OA is rarely associated with sensorineural deafness and vestibular dysfunction. Inheritance is autosomal dominant. The presence of heterochromia iridis and a prominent white forelock in some patients suggests overlap with Waardenburg
syndrome. Therefore, a digenic interaction between micropthalmia-associated transcription factor (MITF), a transcription factor linked to Waardenburg syndrome, and OA has been proposed (
10). OA is associated with late-onset deafness and X-linked inheritance. This phenotype is attributed to a variant of a G-protein-coupled receptor or contiguous gene defect (
11).
OCA can appear as part of a multisystem disease in which the melanocyte and other intracellular organelles are affected. The classic example is Hermansky-Pudlak syndrome (HPS), which includes a group of genetic disorders resulting from defects in membrane trafficking. Membrane and secretory proteins are synthesized in the endoplasmic reticulum, then move on to the Golgi complex where they undergo posttranslational modifications. These proteins are then shuttled to the plasma membrane, secretory granules or vesicles, and to organelles of the endocytic pathway. To date, eight genotypes have been identified, HPS1-8 (
12,
13,
14). The gene for type 2 HPS encodes for the β3A subunit of adaptor complex 3 is known to assist in the transport of tyrosinase into melanosomes and lytic granules into NK cells and cytotoxic lymphocytes (
15). The remaining HPS proteins are involved in the assembly of additional lysosomal-related organelles. Progressive accumulation of ceroid lipofuscin leads to interstitial fibrosis and granulomatous colitis. A bleeding diathesis can occur as a result of a deficiency of platelet storage granules or dense bodies. Reduction or absence of these granules, which contain serotonin, adenine nucleotides, and calcium, results in defective platelet aggregation. Easy bruisability, of soft tissues especially, and prolonged bleeding time in individuals of Puerto Rican descent are characteristic features.
Chédiak-Higashi is another multisystem disease associated with albinism. Patients with this disorder have the typical features of albinism, and skin biopsy reveals the presence of abnormally large melanosomes. In addition, they have an associated immune defect, increased susceptibility to lymphoproliferative disorders, and presence of larger intracytoplasmic granules in leukocytes and other tissues. Defective chemotaxis and decreased bactericidal activity predisposes these children to bacterial infections. The diagnosis is usually suspected on the basis of the clinical findings and presence of abnormal granules in their leukocytes. Most patients die by the second decade of life from an overwhelming infection or malignancy.
Cystinosis
Cystinosis is an autosomal recessive disorder with an estimated incidence of 1 case per 150,000 live births. It is a lysosomal storage disorder resulting from defective transport of cystine across lysosomal membranes (
20). Cystine is the homodimer of the amino acid cysteine, which is generated by protein catabolism. Cystinosin, a selective transmembrane protein, transports cystine out of the lysosome. Mutations in the gene encoding for cystinosin (
CTNS) impair cystine transport, resulting in accumulation at levels of 5 to 500 times normal—levels that initiate crystallization and cell damage (
21). The most common mutation is a 57,257 bp deletion that can be detected by polymerase chain reaction (
22). The kidney is particularly susceptible to cystine toxicity, resulting in kidney damage beginning in the first year of life. By comparison, central nervous system (CNS) damage does not become evident before the patient’s third decade of life (
23).
The term
cystinosis includes infantile and late-onset nephropathic forms and a benign nonnephropathic form. The most common and clinically important type is the infantile nephropathic form. Infants with cystinosis are typically normal at birth. Beginning at about 6 months of age, development slows, linear growth falls, and the child suffers from isolated or repeated episodes of acidosis and dehydration. Urinalysis reveals excessive losses of glucose, amino acids, phosphate, calcium, bicarbonate, and other small molecules (Fanconi syndrome). Progressive glomerular damage usually leads to renal failure in untreated patients by 10 years of age (
29). Phosphaturia can lead to hypophosphatemic rickets. Accumulation of cystine crystals in other tissues causes hypothyroidism, diabetes mellitus, and delayed onset of puberty. Patients with treated nephropathic cystinosis can develop late complications such as distal myopathy, swallowing difficulties, hepatomegaly, and cortical atrophy and calcifications evidenced by computed tomography (CT) (
23).
Late-onset cystinosis is clinically similar to the infantile form except for the delayed onset and slower progression of the disease. Patients may have preserved renal function into the third decade of life, and growth delay is mild. Accumulation of corneal crystals is slower. Patients with nonnephropathic disease have isolated ocular involvement. Molecular testing reveals the presence of mutations with residual activity of the cystine transporter (
24).
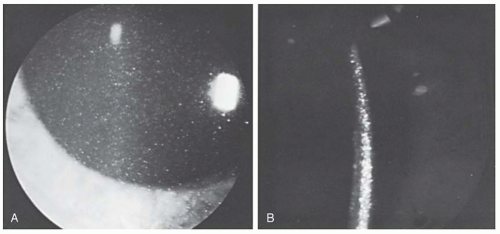
The eye, like the kidney, is predisposed to cystinerelated damage. Although they are not present at birth, cystine crystals can be found in the cornea of patients with nephropathic cystinosis by 1 year of age (
Table 21.2). Slit-lamp detection of corneal crystals is an easy way to help confirm the diagnosis and should be done in all patients with Fanconi syndrome or renal failure of uncertain etiology. These refractile crystals first appear in the epithelium and anterior stroma of the cornea, but with increasing age, they occupy its entire thickness and become densely packed (
Fig. 21.3). With continued buildup of cystine crystals, the cornea becomes opacified, resulting in decreased vision and the need for corneal transplantation in some patients. Cystine crystals are abundant on the conjunctiva, but there are relatively fewer on the surface of the iris, lens capsule, and trabecular meshwork.
Photophobia and secondary blepharospasm are significant problems for most patients with cystinosis and can be disabling in some patients. Symptomatic children often wear heavily tinted sunglasses and brimmed hats to avoid undue levels of light
exposure. The severity of photophobia seems to be correlated with the density of cystine crystals rather than the loss of the corneal epithelium. Possible reasons for the photophobia include a tear deficiency, subtle corneal inflammation, and glare (
25).
Increased long-term survival of patients with cystinosis has led to the emergence of late ocular sequelae (
26). The most important sequelae are due to progressive cystine accumulation in the retina (
27). Decreases in visual acuity, color vision loss, and pigmentary disturbances of the macula and peripheral retina have been described (
Table 21.3). Visual testing shows elevation of dark-adaptation thresholds, visual field constriction and variable reduction in the rod- and cone-mediated ERGs. Severe visual loss and even blindness can occur in patients who go untreated for years. Progressively increasing deposits of cystine crystal can be detected in most ocular tissues, including extraocular muscles and the optic nerve.
Replacement of electrolytes, calcium, carnitine, and glucose due to renal losses, kidney transplantation, and oral cysteamine are the mainstays of systemic treatment. Several studies have documented the ability of cysteamine to preserve renal function and to prevent growth retardation (
28,
29). However, cystine crystals continue to accumulate in the cornea, indicating that the drug does not achieve adequate levels in this avascular tissue. To overcome this delivery problem, topical cysteamine was formulated and found to be highly successful in removing cystine crystals from the cornea. Cysteamine eye drops (0.5%) administered as often as hourly are well tolerated and dramatically reduce the intense photophobia and blepharospasm that accompany cystine accumulation (
30).
Hyperornithinemia (Gyrate Atrophy of the Choroid and Retina)
Gyrate atrophy of the choroid and retina is a retinal degeneration related to a deficiency of ornithine aminotransferase (OAT). As a result, 10- to 20-fold elevations of ornithine accumulate in plasma, cerebrospinal fluid (CSF), and aqueous humor. More than 50 different mutations of the gene encoding for OAT have been identified to date (
31). The functional gene has been mapped to chromosome 10q26. Gyrate atrophy occurs worldwide, but its prevalence is much higher in the Finnish population.
Clinically, the disease begins with myopia, night blindness, and contracted visual fields in the first decade of life (
32). Examination of the fundus reveals punctate or circular patches of chorioretinal atrophy in the retinal periphery (see
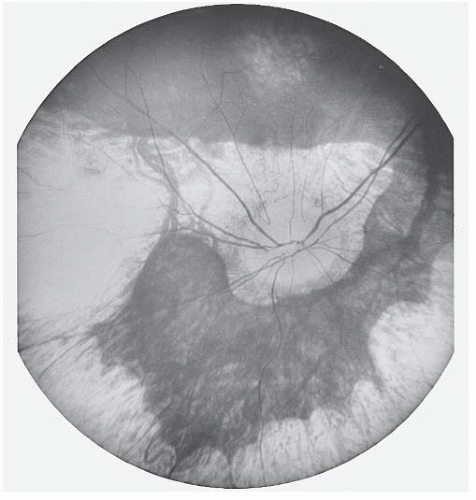
Table 21.3). With increasing age, the lesions become larger and more numerous, gradually coalescing and forming confluent areas of chorioretinal atrophy with scalloped margins posteriorly (
Fig. 21.4). Near puberty, increased pigment appears at the back edge of the chorioretinal lesions and in the posterior pole. Posterior subcapsular cataracts are present in patients by the end of their second decade. Visual loss parallels the fundus changes, showing slowly progressive contraction of the visual field until blindness occurs in the third to seventh decades of life. Likewise, the ERG progressively deteriorates until the response to light becomes extinguished.
Various therapies have been tried to prevent or delay the onset of blindness. Initially pharmacologic doses of vitamin B6, a cofactor for OAT activity, were given in an attempt to stimulate the enzyme. This strategy failed in all but a small percentage of patients (
33,
34). A second strategy involved creatine supplementation based on the fact that ornithine is a potent inhibitor of creatine biosynthesis. Creatine supplementation appeared to have no beneficial effect (
35).
Another strategy called for an arginine-restricted diet with the intent of lowering plasma ornithine levels. The effect of long-term reduction of ornithine accumulation is still controversial. Some authors report slowed progression of the retinal degeneration, while others observe continued progression (
36,
37,
38).
Primary Hyperoxaluria
Primary hyperoxaluria (PH) is a rare autosomal recessive disorder characterized by increased synthesis and toxic accumulation of oxalic acid. Failure of either of two enzymes to detoxify glyoxalate, an oxidation product of glycine, leads to its increased conversion to oxalate. PH type 1 (PH1), which is the most common type, is due to a deficiency of the liverspecific peroxisomal enzyme alanineglyoxylate aminotransferase (
39). PH type 2 (PH2) is caused by a defect in the more widespread cytosolic enzyme D-glycerate dehydrogenase/glyoxylate reductase (
40). Because oxalate cannot be metabolized further, elevated levels accumulate in the urine, leading to the formation of stones and resulting in nephrocalcinosis in the kidney, and eventually renal failure. In PH1, oxalate crystals can accumulate in extrarenal tissues including bone, heart, nerves, joint, and teeth (oxalosis). In general, PH2 is a milder disease, and there is no evidence of systemic oxalosis.
Retinopathy is the major clinical finding in the eye, but histopathologically oxalate crystals are deposited throughout the ocular tissues (
41,
42). Initially, multiple crystalline deposits (100 to 200 µm) with a vascular distribution are found in the posterior pole out to the equator. Later, ringlets of pigmentation may surround the crystals and coalesce in the macula, forming a black geographic lesion (see
Table 21.3). Visual acuity is relatively good when the optic disk is normal but reduced when there is optic atrophy (
42). Fluorescein angiography initially shows multiple areas of hypofluorescent centers (oxalate crystal) surrounded by hyperfluorescent rings (atrophic RPE). Later, angiography can show small-vessel occlusion and localized subretinal neovascularization.
The diagnosis is usually based on the presence of increased urinary excretion of oxalate and glyoxylate in PH1, and of oxalate and glycerate in PH2. Enzyme assay of biopsied tissue or molecular testing is necessary to confirm the diagnosis. Treatment is directed at reducing exogenous oxalate intake, administration of pharmacologic doses of pyridoxine (essential cofactor for aminotransferases), or enzyme replacement by liver transplantation. The associated renal failure is managed in the short term with renal dialysis and in the long term with kidney transplantation (
43).
Galactosemia
The galactosemias are a group of three inherited disorders characterized by an inability to metabolize galactose. The main source of galactose is milk, which contains lactose, a disaccharide composed of glucose and galactose. Galactose is primarily converted to glucose by sequential enzymatic activity of galactokinase, galactose-1-phosphate uridyl transferase (Gal-1-UDP transferase), and uridine diphosphate (UDP)-galactose-4-epimerase. Decreased activity of any one of these enzymes is a cause of galactosemia. The gene for each galactose enzyme has been characterized, and numerous mutations have been identified (
44,
45,
46).
Patients with Gal-1-UDP transferase deficiency, the most common form of galactosemia, typically present in infancy with failure to thrive. Vomiting and diarrhea begin with milk ingestion. Jaundice and unconjugated hyperbilirubinemia are the earliest signs of hepatic dysfunction, followed by hepatomegaly and abnormal liver function tests. Untreated, the liver disease can progress to cirrhosis. Aminoaciduria and proteinuria are evidence of renal tubular dysfunction, and there is a higher incidence of
Escherichia coli sepsis. A minority of patients present with developmental delay, hepatomegaly, and cataracts later in life (
47).
Cataract is the major ocular complication (
Table 21.4). The cause of the cataract is most likely related to the
accumulation of galactitol in the lens. In the presence of lenticular aldose reductase, galactose is reduced to galactitol, which is impermeable to cellular membranes, leading to its intracellular accumulation. Increased intracellular levels of galactitol create an osmotic gradient causing the lens to imbibe water, which in turn leads to hydropic degeneration of lens fiber cells. The earliest observable lens change is an increased refractive power of the fetal lens nucleus, giving the appearance of an “oil drop.” This is usually followed by development of a zonular or nuclear cataract (
48).
The mainstay of treatment is elimination of galactoserich foods from the diet. Long-term studies indicate that dietary elimination of galactose can reverse or delay the development of cataracts and liver disease, but it has no ameliorative effect on the damage to the CNS and ovaries (
49). Children with galactosemia are often delayed in acquisition of language skills, mildly retarded, and girls suffer from ovarian failure.
Patients with galactokinase deficiency have cataracts but there is no evidence of hepatic or renal disease, or mental retardation (
50). Because cataracts may be the first and only abnormality, it is important to routinely screen for galactosemia in infants and children who develop cataracts (
51). Homozygous deficiency of galactokinase is associated with the zonular cataracts that typically develop in the first year of life. By comparison, the causal relationship between partial galactokinase deficiency and cataracts is less clear. Individuals with reduced galactokinase activity seem to have a higher prevalence of cataracts than those with normal galactokinase activity. Cataracts that develop later in life can be nuclear or subcapsular. Rarely pseudotumor cerebri has been reported.
There are two forms of the UDP-galactose-4-epimerase deficiency. In the benign form, the child is normal and the epimerase deficiency is limited to red blood cells and leukocytes. With generalized loss of epimerase activity, the clinical presentation resembles transferase deficiency with vomiting, weight loss, hepatomegaly, hypotonia, aminoaciduria, and galactosuria. Cataracts have not been noted in either form of this rare disorder (
49,
52).