Nutritional Blindness: Xerophthalmia and Keratomalacia
Alfred Sommer
Xerophthalmia and keratomalacia are the most common, most devastating ocular diseases attributable to nutritional deficiency. In general, nutritional blindness refers specifically to these two interrelated conditions.1,2,3
Although xerophthalmia literally means “dry eye,” it denotes the entire spectrum of ocular abnormalities arising from vitamin A deficiency. These include night blindness, retinopathy, conjunctival and corneal xerosis, corneal ulceration and melting, and less obvious alterations in the epithelial structure of the eye and several other organs.
Keratomalacia, which is less well understood and defined, is commonly described as “liquefactive” or “colliquative” corneal necrosis. Often the last, most severe stage of xerophthalmia, it also occurs in other conditions, such as severe measles, in which general malnutrition usually is present but specific signs of vitamin A deficiency may be absent. These latter situations may represent acute decompensation of vitamin A status, secondary herpes or other infections, or the destructive effects of traditional medicines.2,3,4
Although the ocular complications of deficiency are best known and clinically evident, they represent only one facet of this multisystemic disorder. The importance of vitamin A for normal host resistance and survival has become increasingly evident. Even children with mild, subclinical deficiency are at increased risk of respiratory disease, diarrhea, anemia, growth retardation, and death.3,5,6
ETIOLOGY, PATHOGENESIS, AND PATHOLOGY
Night blindness and its cure were recognized in pharaonic Egypt more than 3,500 years ago. In the late 19th century, two Frenchmen, Hubbenet and Bitot, described the association between localized conjunctival xerosis and night blindness, and an Englishman, Snell,7 demonstrated that both conditions respond rapidly to administration of cod liver oil. In the early 20th century, Bloch, a Danish pediatrician, elucidated the relation between these milder aberrations, blinding corneal destruction, and a diet deficient in the then newly discovered fat-soluble vitamin A.8 There is now little question that vitamin A deficiency is responsible for all xerophthalmic manifestations, from night blindness through corneal xerosis, and for most forms of xerophthalmic corneal ulceration.1,2,3,9,10
Vitamin A is a fat-soluble product of animal metabolism found principally in the liver of fish, poultry, and livestock, and in eggs and dairy products. Carotene, especially beta carotene, present in certain orange and yellow fruits and in dark green leafy vegetables, is converted in the gut to vitamin A. Because of inefficiency in this conversion, 12 to 28 times as much beta carotene is needed from dietary foods as is preformed vitamin A for the same effect.3,11,12 After absorption, approximately 50% of the vitamin is retained and stored in the liver. These liver stores form an important buffer against fluctuations in vitamin A intake. When needed, vitamin A is released into the bloodstream in association with retinol binding protein (RBP), a specific carrier protein elaborated by the liver. The associated complex is known as holo-RBP. Retinol is required for the elaboration of rhodopsin or “visual purple” by the retinal rod cells, for maintenance of normal differentiation of the epithelial lining of several structures throughout the body, and for a fully competent immune response.3
Conditions that interfere with ingestion, absorption, storage, or transport of vitamin A may precipitate xerophthalmia. The most common is inadequate dietary intake. Most vitamin A deficiency in the developing world,1,13 and even an occasional case in developed countries, can be traced to inadequate diet. Recommended daily allowances for vitamin A range from 300 to 1,200 μg of retinol or its equivalent.1,14 When intake falls much below this amount, liver stores are drained to maintain serum retinol at a normal level. Chronic deficiency results in depletion of liver stores, a fall in serum retinol levels, and the appearance of clinical disease. At the same time, RBP accumulates in the liver. An increase in vitamin A intake will result in a massive outpouring of holo-RBP from the liver and rapid rise in serum holo-RBP levels.15
Lack of dietary lipid, impaired secretion of digestive enzymes (as in cystic fibrosis16), gastroenteritis, celiac sprue,17 and heavy worm loads all interfere with the absorption of ingested vitamin A. This has the same effect as reduced vitamin A consumption.
Severe protein deficiency interferes with protein synthesis, thus reducing vitamin A absorptive, storage, and, most importantly, transport capacities. The serum level of holo-RBP declines, as does the concentration of RBP in the liver. If vitamin A intake remains high, liver stores of retinol may rise.18 Increased protein intake results in a substantial rise in circulating vitamin A.19 Whether isolated protein deficiency can interfere with vitamin A transport to a clinically significant degree is still uncertain but appears likely.10
These conditions are exacerbated by febrile illnesses, which increase metabolic demands for what little vitamin A is available. Persons already having borderline vitamin A status, but without frank disease, can lose vision in both eyes within a few days of the onset of measles, severe gastroenteritis, or bronchopneumonia.2,3,4
Serum vitamin A levels in well-fed western populations are generally between 30 and 40 μg/dL. Although impaired scotopic vision has been recorded at levels above 30 μg/dL,20 clinical disease is exceedingly uncommon at levels above 20 μg/dL,2,3 though individuals probably suffer from impaired immune responsiveness and may be more susceptible to severe infections and other nonocular physiologic disturbances. Approximately 10% of children with levels between 10 and 19 μg/dL will have night blindness, conjunctival xerosis, or both. Between 10% and 50% will have epithelial metaplasia detectable by impression cytologic study. The proportion of persons with clinical disease, especially the more severe forms of corneal involvement, rises appreciably at lower vitamin A levels, especially at those below 10 μg/dL.2,9
Night blindness is the best understood of all xerophthalmic changes. Vitamin A is an essential component of rhodopsin, the pigment responsible for rod function.21 Although most vitamin A liberated during bleaching is recycled, a small proportion is continually lost and must be replenished by fresh supplies. When these are unavailable, the retina fails to attain its normal, dark-adapted sensitivity. Severe, prolonged deficiency of vitamin A, in the rat at least, results in degeneration of retinal pigment epithelium and photoreceptors.22 Clinical data suggest similar changes may occur in humans.23
Epithelial changes consequent to vitamin A deficiency are equally well recognized but less well understood. Vitamin A is required for maintenance of normal epithelial architecture, particularly of mucous membranes. In its absence, keratinizing metaplasia occurs in a variety of organs.24 The conjunctiva, because of its accessibility, has been the best studied.23,25,26 Goblet cells are lost, and keratinized material, often containing numerous saprophytic organisms (especially the xerosis Bacillus, a gram-positive diphtheroid), covers the surface (Fig. 1). Below the keratinized surface is a thickened layer of flattened cells and, deep to this, a prominent granular cell layer. The architecture of deeper layers is moderately disorganized. A mild, chronic inflammatory infiltrate may be present in the substantia propria. Occasionally, in severe cases, rete pegs are present.27
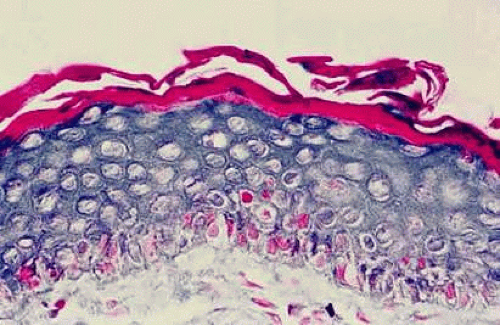 Fig. 1 Histopathologic changes of conjunctival xerosis in a 25-year-old Indonesian woman. The epithelium is characterized by keratinization, a prominent granular cell layer, and distended squamous cells with large, open nuclei and prominent nucleoli. A mild mononuclear cell infiltrate, not apparent in this section, also was present. (Hematoxylin-eosin, 700X.) (From Sommer A, Sugana T, Djunaedi E, Green R: Vitamin A–responsive panocular xerophthalmia in a healthy adult. Arch Ophthalmol 96:1630, 1978, with permission.) |
Histopathologic material of mild corneal changes displays the same flattening and keratinization of epithelial cells.28
Conjunctival and corneal keratinization appear to be primary events, independent of the loss of mucus-producing goblet cells and alterations in tear film dynamics.23,26,29
Keratomalacia represents frank corneal necrosis (Fig. 2), with fragmentation and dissolution of stromal collagen and loss of keratocytes.27,28,29 Its pathogenesis, however, is unclear. Although most cases are accompanied by clinical xerophthalmia, African children develop a similar picture after measles, often in the absence of overt signs of vitamin A deficiency. Since measles does not produce corneal necrosis among well-nourished western or African children, vitamin A deficiency, protein deficiency, and as yet undetermined factors have been suspected.4,30 Studies from Nigeria and Tanzania suggest herpes virus infection may play a role in numerous cases, whereas diagnostic, laboratory, and clinical studies indicate vitamin A deficiency is responsible for half of all resultant blindness.4,31
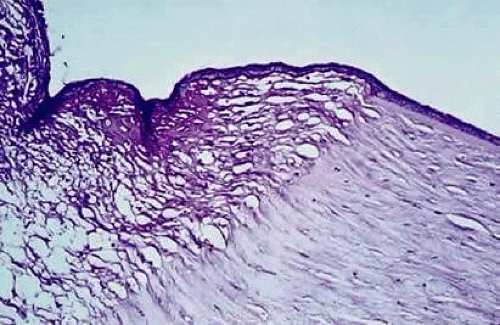 Fig. 2 Characteristic changes of keratomalacia in autopsy specimen from a severely marasmic 1-year-old Indonesian boy with bilateral corneal ulceration, a serum vitamin A level of only 10 μg/dL, and a holo retinol binding protein value of only 2 μg/mL. The sharply demarcated area of stromal necrosis is covered by keratinized epithelium. There is little inflammatory infiltrate and no bacteria in the area of collagen dissolution. (From Sommer A, Green WR, Kenyon KR: Clinical-histopathologic correlation in xerophthalmic ulceration and necrosis. Arch Ophthalmol 100:953, 1982, with permission.) |
NATURAL HISTORY AND EPIDEMIOLOGY
The epidemiology of xerophthalmia is the epidemiology of vitamin A and protein deficiency. Substitution of unfortified margarine for dairy products in turn-of-the-century Denmark,32 or sweetened condensed milk for breast-feeding in pre–World War II Singapore were accompanied by epidemics of xerophthalmia. Appreciation of the causes of xerophthalmia and changes in public policy have almost eliminated nutritional blindness from these and other developed countries. Nonetheless, 140 million children and over 7 million pregnant women are thought to be physiologically deficient every year, and xerophthalmia remains a worldwide problem of enormous magnitude, especially in underdeveloped regions of Asia, where the diet consists mainly of rice, and in Africa.3,4,33,34,35,36,37 Moderate numbers of cases also occur in the Caribbean38 and in Central39 and South America.40 On the basis of data from Indonesia,41 it is estimated that as many as 5 to 10 million Asian children develop clinical xerophthalmia every year, and of these, 5% to 10% have corneal involvement (one fourth to one half of whom go blind). Overall mortality rate in untreated cases of corneal xerophthalmia is probably 50% to 90%. Children with mild xerophthalmia (night blindness, Bitot spots, or both) die at three to nine times the rate of their better-nourished peers.42
In xerophthalmia-endemic developing countries, the disease is largely confined to lower socioeconomic groups. For the most part, these groups cannot afford vitamin A–rich foods, and the most palatable sources of beta carotene, such as papaya and mango, are sold as cash crops. Green leafy vegetables are avoided by young children, whereas the tenacity with which their beta carotene is bound to cellular matrices may preclude their providing adequate vitamin A nutriture.3,11,12
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


