Chapter 96 Nonrhegmatogenous Retinal Detachment
 For additional online content visit http://ww.expertconsult.com
For additional online content visit http://ww.expertconsult.com
Introduction
A wide variety of diseases may present with sensory retinal detachment without retinal breaks. Nonrhegmatogenous retinal detachment may be exudative in nature or caused by vitreoretinal traction. Some diseases with elevated retina may have both exudative and traction components. In exudative retinal detachment, the subretinal fluid may be confined to a localized area, usually the posterior pole, or may extend to the periphery, even forming bullous retinal detachment. The characteristic feature of a significant exudative retinal detachment is the presence of shifting subretinal fluid.1 The fluid shifts to the most dependent location when patients change body position. The surface of the detached retina is usually smooth; however, retinal folding may occur in some diseases associated with subretinal fibrosis. To reach an accurate diagnosis among many diseases presenting with exudative retinal detachment, careful fundus examination, fluorescein angiography (FA), indocyanine green angiography (ICGA), optical coherence tomography (OCT), ultrasonography, computer tomography (CT), and magnetic resonance imaging (MRI) may be necessary.
Idiopathic
Central serous chorioretinopathy
Bullous retinal detachment
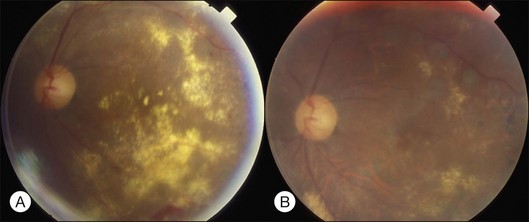
Bullous RD usually has an acute onset with simultaneous or sequential involvement of the two eyes. Fundus examinations reveal multiple areas of serous RD in the posterior retina with lower bullous RD. There may be multiple retinal pigment epithelial detachment (RPED) and one or more grayish or yellow patches of subretinal exudates mimicking focal chorioretinitis (Fig. 96.1). In some cases, retinal folds may form by the contraction of fibrinous patches or fibrotic membrane or bands on the outer surface of the detached retina. Small scattered yellowish granular subretinal deposits may have a tendency to settle along the retinal vessels (Fig. 96.2). The vitreous is usually clear, but may have 1–2 plus cells. The disc is not hyperemic.
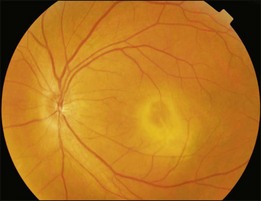
Fig. 96.1 Subretinal fibrin-like deposition mimicking chorioretinitis in a case of acute bullous detachment.
Optical coherence tomography may show retinal pigment epithelial detachment with or without sensory detachment adjacent to or overlying it; the subretinal fluid may be clear or slightly turbid with multiple granular deposits above RPE and on the outer surface of the detached retina, sometimes forming incomplete septa within the subretinal space (Fig. 96.3); subretinal fibrinous mount with surrounding sensory detachment may be seen.2
Complications of bullous RD include: large RPE tear; broad retinal folding; submacular plaques or fibrotic bands; peripheral paravascular exudates; peripheral retinal telangiectasia, occlusion, or even fibrovascular proliferation.2,3 Peripheral vascular changes may be secondary to long-standing sensory detachment.
In treating bullous RD, systemic steroids should be discontinued; patients should keep the head elevated during sleep to prevent fluid shifting to the macular area; FA-guided laser to the leaking points may decrease the subretinal fluid. Once the fluid level recedes, FA should be repeated to identify persistent leaking points and other leaking sites previously hidden by the detached retina. Multiple sessions of laser treatment are usually needed for complete fluid reabsorption. If subretinal fluid (SRF) persists after the above measurements, external drainage of SRF may be undertaken.4 Care should be taken to make sure that the surgical drainage site is posterior enough to access the subretinal space, which is in the dependent area. Alternatively, pars plana vitrectomy with perfluorocarbon liquid injection and simultaneous external drainage through anterior sclerotomy may be performed, followed by focal laser to the exposed leaking sites and air–fluid exchange. The effect of vitrectomy with internal drainage and silicone oil tamponade is controversial. Recently, bevacizumab injection has been shown to rapidly reduce active fluid leakage into the subretinal space as well as decrease the deposition of fibrinous or proteinaceous substances.5 Photodynamic therapy (PDT) with reduced fluence may also reduce choroidal hyperpermeability and facilitate subretinal fluid reabsorption with RPE tear being the major complication.6 Prognosis of bullous RD is variable and is affected by the duration of macular detachment, the presence of submacular fibrosis, the development of submacular RPE tears, and occurrence of fibrovascular proliferation under the macula or in the periphery.
Chronic CSCR
Typical clinical manifestation is the multiple poorly defined areas of chronic persistent or recurrent retinal detachment in the posterior pole. Subtle or obvious areas of RPE changes are noted in the posterior pole and in the juxtapapillary regions; gravity tract forming vertical band or reverse funnel-shaped depigmentation, along with pigment migration or bone-spicule pattern of pigmentary changes within the tract and in the inferior part of the retina are usually found (Fig. 96.4); shallow or bullous detachment in the inferior retina is a frequent finding.
Photocoagulation remains the main treatment method. Conventional laser or the more recently developed MicroPulse laser to leaking points and areas of RPE changes with late fluorescein staining and leakage may stop the leakage.7 PDT with reduced fluence has been advocated to treat leaking points and areas with late oozing shown in FA with good effect. Intravitreal bevacizumab has also been shown to have therapeutic effect.8 However, the prognosis is guarded because of multiple recurrence and permanent macular RPE disturbance.
Uveal effusion syndrome
Histopathological examination shows accumulation of protein-rich extracellular materials in the suprachoroidal and subretinal spaces. Choroidal vessels are dilated without inflammatory cell infiltration. Subarachnoid space around the optic disc is enlarged. The sclera shows deranged fibers with deposition of glycosaminoglycans within.9 Cell culture of the scleral cells reveals intracellular deposition of a glycogen-like substance.10
The pathogenesis is unclear, possibly related to congenital anomaly of the sclera and vortex veins hypoplasia. Excessive glycosaminoglycans accumulate within the sclera combined with defective vortex veins resulting in decreased drainage of extravasated protein through scleral emissary channels of the transscleral outflow pathway; fluid drainage is also compromised from the decreased function of uveoscleral outflow pathways, leading to excessive protein and fluid accumulation in the suprachoroidal space. Later on, the protein and fluid enter the subretinal space when the extracellular protein concentration becomes equal to that within the vessels. Protein in the suprachoroidal space around the disc may gain access to the subarachnoid space and subdural space resulting in an increase in the CSF protein content even without pleocytosis in 50% of the patients.11 Forrester and associates believe that IUES is a kind of ocular mucopolysaccharidosis, with the initial defect resting in the proteodermatan synthesis and/or degradation of the fibroblast of the sclera.12 Other evidence also shows that abnormal mucopolysaccharides of the sclera play an important role in the pathogenesis of IUES. IOP is usually within normal limits because the IOP rising tendency from uveoscleral outflow obstruction is neutralized by decreased aqueous production from the ciliochoroidal detachment.
Best treatment methods have been debated. Vortex vein decompression with scleral resection was initially advocated to treat uveal effusion associated with nanophthalmos. Gass believed that the treatment effect had less to do with vortex vein decompression than with scleral resection to facilitate protein and fluid drainage through the sclera.13 He suggested partial-thickness sclerectomies or full-thickness sclerectomies. After treatment, exudation may gradually disappear within a few months. However, chorioretinal degeneration may continue to develop from chronic mild recurrence of exudation or abnormal metabolism of mucopolysaccharide.
Vascular
Coats disease
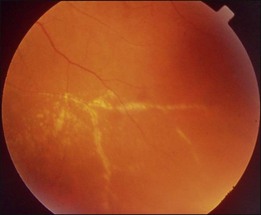
This is a non-familial developmental retinal vasculopathy. The disease is more common in males, is usually unilaterally affected and may occur in infants. Symptoms often develop in children or young adults; one-third had symptoms onset over 30 years of age. All vessels, arteries and veins alike, would be affected, showing telangiectasis combined with a large amount of hard exudates; hemorrhagic retinopathy is occasionally seen. On the other hand, a minor form of the disease mainly involving juxtafoveolar areas may occur; decreased vision will not occur until adulthood when hard exudates and edema develop in the macula. The prognosis is directly influenced by the size of the involved area. Occasionally, other vascular anomalies may appear in the lesion eye or the fellow eye, such as macular macrovessels or arterial tortuosity (Fig. 96.5). The condition may be occasionally associated with other abnormalities such as progressive facial hemiatrophy, facial scapulohumeral muscular dystrophy and deafness, or Alport syndrome.14 It may rarely accompany systemic vascular anomalies.
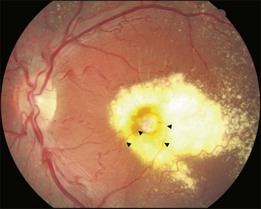
Fundus examinations reveal changes of various severities. The mild form presents with focal telangiectasia and microaneurysms usually at the temporal side of the macula, with or without mild hard exudates. The moderate form ranges from cystoid macular edema with significant hard exudates surrounding the area containing telangiectatic vessels or microaneurysms, to the more extensive vascular abnormalities with massive exudates which may gain access to the subretinal space. The severe form shows wide and scattered vascular lesions, with hard exudates accumulating around the disc and in the posterior pole, causing exudative detachment. The macula may be detached with massive intra- and subretinal exudates, which later may transform to organized subretinal disciform mass or atrophic scar. These changes are likely to be found in infants and children, who visit the ophthalmic clinic because of manifest strabismus secondary to unilateral poor vision or abnormal red reflex from massive exudates in the posterior pole. The accumulation of exudates in the macula may be due to gravity-induced migration of subretinal exudates toward the central area during sleep. The deposition of lipid-rich substance along with macrophage evolves into fibrous tissue. Retinal or choroidal vessels may grow into the lesion to form a disciform scar. The most advanced form presents with bullous detachment with the retina coming in direct contact with the crystalline lens; cholesterol crystals accumulate in the subretinal space.15 There may be dilated abnormal vessels, hard exudates or hemorrhage on the surface or in the retina. However, the abnormal tortuous vessels do not dip into the subretinal space, a sign typical of exophytic retinoblastoma.
Treatment usually involves laser or cryo aiming at the lesions to decrease exudates and preserve vision (Fig. 96.6). For severe exudative detachment, external drainage should be performed first, followed by cryo to the abnormal vessels. Scleral buckling may facilitate retina reattachment, enhance cryo effect and promote regression of abnormal vessels. However, new lesions may develop in nearby or remote areas. Follow-up is crucial to detect and treat new lesions. Sector panretinal photocoagulation (PRP) may be used for a nonperfusion area. For severe cases, vitrectomy to release vitreous traction with external subretinal fluid drainage, laser or cryo may be considered. Recently, repeated intravitreal injection of bevacizumab has been reported to reduce subretinal fluid, facilitating subsequent laser or cryo.16
Accelerated hypertension and pregnancy-induced hypertension
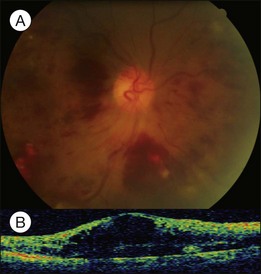
While chronic moderate hypertension is rarely associated with choroidopathy, choroidal ischemia is more frequently associated with accelerated hypertension. Unlike retinal circulation, choroidal vessels do not possess autoregulation; the blood flow during fluctuation of systemic blood pressure is mainly regulated by sympathetic tone. When blood pressure is high, raised sympathetic tone can prevent direct pressure damage to the choriocapillaries; however, if there is a rapid rise in blood pressure, excessively increased sympathetic tone may prompt severe constriction of choroidal arteries and arterioles, leading to ischemic changes of the choriocapillaries. Choroidopathy may be separated into three stages: (1) acute ischemic phase; (2) chronic occlusive phase; (3) chronic reparative phase.17 In the first two phases, fundus examination may observe white or reddish patches in the outer retina, possibly caused by RPE necrosis; exudative detachment is often present. FA may show a large confluent area or scattered areas of choroidal filling delay; in the mid and late phases, multiple dots or a mosaic pattern of hyperfluorescence from RPE leakage may be seen. In the reparative phase, large areas of irregular REP atrophy, Elschnig spots (central hyperpigmented and peripheral hypopigmented lesion of RPE changes), or Siegrist spots (spots of pigmentary changes similar to Elschnig spots arranged linearly along choroidal vessels in the equatorial region) may be seen; exudative detachment disappears, but choroidal delayed filling remains.
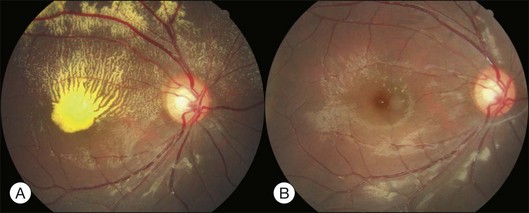
Pregnancy-induced hypertension
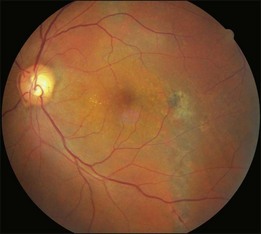
About 1–2% of pregnant women develop exudative retinal detachment pre- or immediately post-delivery, causing visual impairment. Exudative detachment may be limited to the macular area or appear as bullous detachment. The retina may or may not show cotton-wool patches or other changes secondary to hypertension retinopathy (Fig. 96.7). Yellowish-white patches of RPE necrosis may be seen. FA shows delayed choroidal filling and multiple leaking points where RPE has been damaged. After delivery, with control of hypertension, exudative detachment rapidly subsides. Most patients have good visual recovery. The posterior pole may show RPE changes forming hyperpigmented lines or patches mixed with yellow spots of RPE atrophy. The bilateral changes may be mistaken for macular dystrophy. Severe cases may have extensive exudative detachment. Widespread RPE changes similar to tapetoretinal dystrophy and severely compromised vision may result. The cause of chorioretinal changes in pregnancy-induced hypertension is not clear. Blood pressure may not be very high before the onset of exudative detachment. Affected patients may have other symptoms and signs related to disseminated intravascular coagulation, such as hemolysis, low platelet count, and elevation of liver enzymes. It is possible that mechanisms capable of inducing disseminated intravascular coagulation (see below) are also functional in the choroid, causing choroidal ischemia, RPE damage and exudative detachment.
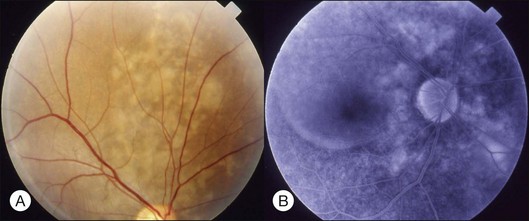
Diabetic retinopathy
Severe diabetic macular edema may sometimes accompany localized macular detachment. Fluid leaking out from the vessels first accumulates within the retina; beyond a certain critical point, fluid may gain access into the subretinal space causing sensory detachment. Severe macular edema not only leads to detachment but may be associated with massive hard exudates (Fig. 96.8). Hard exudates are one of the independent risk factors for vision decrease.18 In addition to vascular hyperpermeability related to diabetic vasculopathy, taut posterior hyaloid membrane may also contribute to macular edema and localized detachment either from the mechanical traction force or from traction-induced increased vascular permeability.
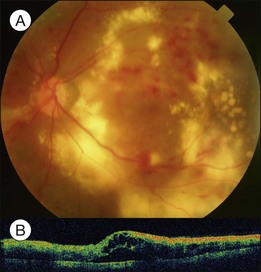
Clinically, early fundus changes leading to severe macular edema may present as a central retinal vein occlusion-like picture with flame-shaped hemorrhage around the disc and scattered perivascular exudates but without fluorescein angiographic evidence of disc leakage and venous delayed filling. In other cases, multiple clusters of microaneurysms may distribute in the posterior pole, accompanied by significant capillary nonperfusion. Sensory detachment and massive exudates may later develop. Exudative detachment combined with macular edema represents severe break down of the inner retinal barrier. Laser alone has little effect in such cases. Multiple sessions of intravitreal anti-VEGF (vascular endothelial growth factors) alone or in combination with subtenon or intravitreal steroid administration may effectively flatten down the retina in most cases. The effect has not been confirmed if subsequent focal laser to the leaking vascular segments or microaneurysms may obtain a more lasting effect. In some cases, exudative detachment is present without significant cystoid macular edema. It may be because the edema is in a resolving phase; thus the response to treatment may be quicker. In severe cases, exudates may consolidate and deposit within or below the macula. If the condition does not improve after several sessions of medical treatment, pars plana vitrectomy combined with hyaloid membrane removal may be performed to reduce edema and hard exudates (Fig. 96.9).19 Because the development of massive hard exudates indicates that the retina is in a relatively hypoxic state or has already gone into the early proliferative stage, panretinal photocoagulation during the operation is required to inhibit production of angiogenic factors, which may induce further macular edema or neovascularization, leading to postoperative vitreous hemorrhage or even neovascular glaucoma.19 In the case of pre-existing posterior vitreous detachment, epiretinal membrane peeling and internal limiting membrane peeling may be considered.20 Massive exudates usually form submacular plaques, affecting vision severely. After surgery, the plaque may reduce in size but does not disappear completely, leaving residual fibrosis or crystal-like deposition, causing permanent decrease of vision. Surgical removal of subretinal exudates through iatrogenic retinotomy has been reported21; the effect has not been firmly established. Patients with severe edema should have a systemic check-up, including blood pressure, blood lipid, and renal function; any abnormalities should be treated, as these may interfere with local response to the treatment.
Vascular occlusive diseases
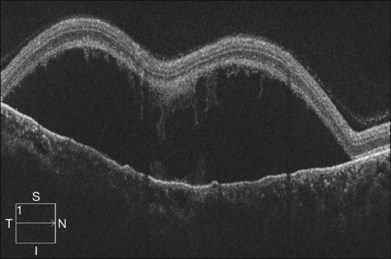
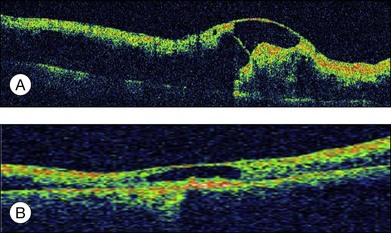
Severe retinal vein occlusion occasionally is accompanied by serous retinal detachment. Exudative detachment has been described in branch, hemispherical, and central retinal vein occlusion (CRVO). Vascular leakage from congested retinal veins outside the macular area is the major source of subretinal fluid at the fovea. In addition, ischemic retina produces angiogenic factors, such as VEGF, which in turn increase vascular permeability. Both increased intravascular pressure and vascular permeability cause leakage of fluid and blood components into the subretinal space. In eyes with retinal vein occlusion, serous RD is typically located beneath the fovea, and the height of the RD was greatest in the fovea (Fig. 96.10).
Recent studies with OCT revealed that macular serous retinal detachment is a common complication of retinal vein occlusion. Serous RD secondary to branch retinal vein occlusion was first described by Spaide et al.22 Of the 14 eyes included in that study, 10 (71.4%) had serous RD. Yamaguchi et al. studied 109 eyes with branch retinal vein occlusion (BRVO) by OCT examination, and found that the incidence of serous RD is higher in the group with major BRVO (63%) than in the group with macular BRVO (21%).23 Ozdemir et al. found a high incidence (81.8%) of serous macular detachment in CRVO.24 In a series of 91 patients with retinal vein occlusion examined by OCT, Tsujikawa et al. reported that 76 eyes (83.5%) had serous RD involving the fovea.25 They suggested from their observations that in eyes with retinal vein occlusion, a small pointed RD developed initially just beneath the fovea, but subsequently changed into a dome-shaped RD; the foveal architecture, especially that of the Müller cell cone might be involved in the formation of serous RD.
Application of laser photocoagulation to the affected area of branch retinal vein occlusion has been reported to treat serous RD and can lead to resolution of subretinal fluid. The beneficial effect of laser treatment may be due to the ablation of ischemic retina, decrease of the production of VEGF, closure of incompetent vessels, and stimulation of RPE to enhance the reabsorption of fluid. Intravitreal injection of triamcinolone or bevacizumab has been reported to treat serous RD due to retinal vein occlusion26,27; repeated injection may be necessary for frequent recurrence of macular edema.
The visual prognosis of serous RD in retinal vein occlusion is variable. In an eye with serous RD associated with retinal vein occlusion, the outer retinal discontinuity does not necessarily lead to poor vision. If the surrounding outer segment of the foveal photoreceptors is preserved, good visual acuity will retain after macular edema and serous RD resolve. However, even after complete resolution of the macular edema and serous RD, diffuse disorganization of the outer photoreceptor layer beneath the fovea often results in poor visual acuity (see Fig. 96.11, online). In addition, a dome-shaped RD sometimes accompanies a focal defect of the outer segment of the photoreceptors above the serous RD. When the defect involves the fovea, visual prognosis is usually poor.
Inflammatory and infectious
Vogt–Koyanagi–Harada syndrome
These associations are high in many populations, including Japanese, Hispanic, Korean, Indian, Italian, Mexican, and Chinese.28
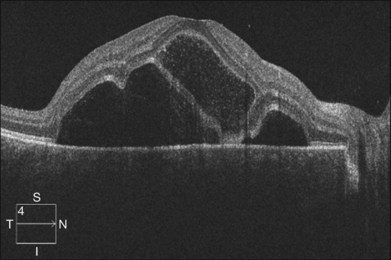
OCT has unique features. In addition to the usual pattern of subretinal fluid accumulation, the subretinal space may develop cystic spaces external to the external limiting membrane (Fig. 96.12); the floors of the cystoid spaces consist mainly of a membranous structure, continuous with the line representing the junction of the photoreceptor inner and outer segments in attached areas. It has been suggested the membranous structures are composed not only of inflammatory products, but also of retinal tissue, probably the outer segment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


