Noninfectious Orbital Inflammatory Disease
James W. Karesh
Alexander V. On
Marc J. Hirschbein
Inflammatory disease involving the orbit represents a diverse group of entities (Table 35-1).
TABLE 35-1. Orbital Inflammatory Processes | |
|---|---|
|
These pathologic processes account for 15% to over 50% of the diseases that occur in the orbit.1,2 Thyroid-related orbitopathy comprises approximately one half of all orbital inflammations. For this reason and for its varied and complex presentation and treatment it is covered separately from the present discussion. Infections account for almost 45% of the remaining inflammatory orbital processes.1,3 The etiologic agents associated with this group and the management of these entities differ significantly from the inflammations to be discussed here. For this reason, they too are described elsewhere.
The inflammatory processes that are the subject of this commentary are for the most part immunogenic and, in general, are idiopathic in origin.4,5,6 The primary exception to this is reactive orbital inflammation secondary to a retained foreign body, nonresorbing blood products, a ruptured dermoid cyst, amyloid deposition, sinusitis, and giant cell reparative granuloma. However, these represent a very small proportion of orbital inflammatory lesions. Of the idiopathic causes of localized orbital inflammation, idiopathic inflammatory orbital disease (IOID), or “pseudotumor” comprises the largest group of nonthyroid noninfectious orbital disease and may constitute between 4.7% and 11% of all orbital lesions.1,4,5,7,8 A separate group of primarily idiopathic inflammatory orbital diseases are those associated with systemic manifestations.4 Many of these are vasculitic in origin, in particular, those associated with collagen vascular diseases and necrotizing granulomatous processes such as Wegener granulomatosis (WG) and idiopathic midline destructive disease. Most of the remaining conditions fall into a group of granulomatous processes of which sarcoidosis is by far the most common.
Orbital inflammation manifests itself in a variety of ways depending upon the acuteness of the disease process. Orbital cellulitis and IOID are most often associated with acute symptomatology, while sarcoidosis demonstrates a more chronic pattern of symptoms. In acute inflammation, symptoms develop over several hours to days. There is usually pain, proptosis, periorbital edema, tenseness, and erythema, as well as conjunctival injection and chemosis. Eyelid ptosis is present secondary to edema. The extraocular motility may be compromised either due to the orbital swelling or direct involvement of the extraocular muscles. Visual acuity may be reduced due to corneal exposure and drying or involvement of the optic nerve either through inflammation or pressure from the surrounding orbital structures. Intraocular inflammation including both uveitis and vitritis as well as retinitis, vasculitis, and papillitis may also be present. Patients may also demonstrate malaise and lassitude. In addition to infectious processes and idiopathic inflammation, aggressive neoplastic diseases such as rhabdomyosarcoma, chloroma, and metastatic tumors, as well as acute hemorrhage associated with lymphangioma or trauma, must also be considered in the differential diagnosis of these findings.
Symptoms and findings associated with subacute inflammation make up the vast majority of manifestations associated with orbital inflammatory disease.3,6,9 These types of symptoms are usually seen in thyroid-related orbitopathy but are also found in IOID, especially myositis dacryoadenitis, WG and other vasculitic processes, chronic infections, particularly those associated with fungi and parasites, and primary or secondary orbital malignancies. In general, subacute processes may either slowly progress over several weeks or months or may have a course that undergoes cycles of progression and remission while worsening. Pain may be present but is usually not as severe as that seen in acute inflammation and may be remitting in character. As with acute inflammatory processes, proptosis, motility disturbances, edema, and injection of the eyelids and conjunctiva are all seen in patients with subacute inflammation. Visual loss and optic disc edema may occur secondary to optic nerve involvement.
Chronic inflammatory orbital disease is usually not associated with significant symptomatology early in its course, although, over an extended period of time proptosis, extraocular motility disturbances, and edema of the eyelids and conjunctiva will usually occur.3,6,9 Pain is often not a feature of chronic inflammation but may occur in sclerosing forms of orbital inflammation. Underlying these findings is a chronic infiltration of the orbital tissues or simply a slowly progressive mass effect. Sarcoidosis, lymphoproliferative and neoplastic processes, thyroid-related orbitopathy, collagen vascular disease, amyloidosis, and reactive granulomatous processes (e.g., cholesterol granuloma and ruptured dermoid cyst) should all be considered in the differential diagnosis.
IOID AND RELATED LOCALIZED INFLAMMATORY CONDITIONS
Idiopathic orbital inflammation is the most commonly reported process associated with orbital inflammatory symptomatology.3,6,9 This is a localized disease process without systemic manifestations. It exists in many variations, the most common of which is known as idiopathic orbital inflammatory disease, formerly known as orbital pseudotumor.5,6 In 1930 Birch-Hirschfeld first used the term pseudotumor to describe orbital inflammation that was associated with proptosis and appeared to be caused by a neoplasm.10 He described three different types of pseudotumor: (i) proptosis that at the time of surgery demonstrates no associated orbital mass, but biopsied orbital tissue shows nonspecific chronic inflammation; (ii) proptosis that at the time of surgery demonstrates an orbital mass, which when biopsied shows nonspecific chronic inflammation, and (iii) proptosis that resolves spontaneously without surgical intervention. However, this does not describe the entire spectrum of manifestations associated with idiopathic orbital inflammation. An evaluation of 65 patients with IOID at Massachusetts Eye and Ear Infirmary revealed the subtypes to include isolated dacryoadenitis 32%, isolated myositis 29%, combined dacryoadenitis and myositis 8%, orbital apex syndrome 9%, and other 22%. The “other: category included involvement of the preseptal region, supraorbital region, sclera, tenon capsule, orbital fat, or optic nerve. The use of imaging modalities allowed further refining of the subtypes by the specific tissues involved, i.e., lacrimal gland, extraocular muscles, orbital fat, preseptal tissues, sclera, episclera, tenons, uvea, optic nerve, orbital mass, and orbital apex and cavernous sinus. IOID can also appear as a diffuse orbital inflammation and may on occasion spread intracranially, cause bony orbital destruction, and rarely, extend into adjacent sinus cavities.6,11,12,13,14,15,16
An important characteristic of all idiopathic inflammatory orbital disease is that no systemic or local cause for the inflammatory response can be identified. In this regard it is a diagnosis of exclusion. The main considerations in the differential diagnosis of this entity are neoplastic processes; thyroid-related orbitopathy; infectious entities such as syphilis, tuberculosis, parasitic infestations, and fungal infections; sarcoidosis; amyloidosis; and various vasculitides including WG and periarteritis nodosa. What remains is a chronic nonspecific inflammatory reaction that is distinctly polymorphic and localized to the orbit. This infiltrate consists of lymphocytes, plasma cells, fibroblasts, macrophages, occasional neutrophils, eosinophils, and epithelial cells; rare granuloma formation and lymphoid follicles; and collagen deposition.3,4,5 The number and composition of these elements may differ with the acuteness or chronicity of the inflammatory process. Similar localized idiopathic inflammatory reactions have been identified in other bodily tissues including the liver, lung, and skin.17,18,19,20,21,22
Epidemiology
As already noted, idiopathic orbital inflammation comprises about 6% of all orbital lesions in various series. No strong sex predilection has been described, except for orbital myositis, which has a 2:1 female predominance. One study reported an overall 1.8:1 female predominance for IOID, with orbital apex syndrome having the highest ratio of 5:1 women to men. There is also no reported racial preference.3 Although it is most common in the third through fifth decades of life, it has been reported in children as well as older adults.23,24,25 It tends to be unilateral, although bilateral disease has been reported and it may even alternate between one orbit and the other.3,26,27 Resistant and recurrent disease occasionally occurs despite appropriate management.
Ocular and Systemic Manifestations
The manifestations of idiopathic orbital inflammation (Figs. 35-1, 35-2, 35-3) vary depending upon which orbital structures are involved.3,6,9 The most common features of this process are pain and proptosis. These symptoms develop rapidly, usually within hours to days, although in some cases they may develop over several weeks. In addition, generally there is chemosis and conjunctival injection often over the extraocular muscle insertions, as well as eyelid edema and ptosis. However, these findings are not always present, and painless, quiet proptosis may occur. Proptosis may not be present in instances of dacryoadenitis.6,9 Instead, eyelid edema and a palpable painful mass in the lateral eyelid may be present. Diplopia and painful disturbances of ocular motility are usually present in the diffuse form of the disease, the superior orbital fissure syndrome, or in myositis but are less common in cases of localized inflammatory masses. Visual loss is unusual but may be associated with intraocular inflammation as well as perineuritis of the optic nerve. Nonspecific constitutional symptoms of malaise and fatigue may also be associated with these various orbital symptoms.17
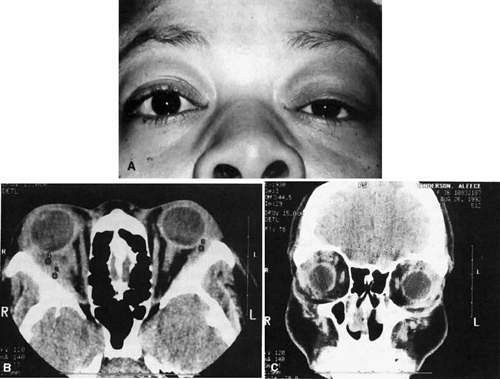 Fig. 35-1. A: A 36-year-old woman who presented with right-sided pain and proptosis occurring over a several-day period. Externally the eye is quiet and does not appear inflamed. The patient had no complaints of diplopia or other visual disturbances. Tissue obtained at biopsy demonstrated idiopathic orbital inflammation. B: Axial CT image from the same patient demonstrates the presence of bilateral orbital masses located laterally in the orbits. Note that masses are ill defined and do not appear to have a capsule. The masses are molded to the bony orbital walls without evidence of any bony irregularities. C: Coronal CT image from the patient again shows the presence of bilateral orbital masses apparently involving the lateral rectus muscles. |
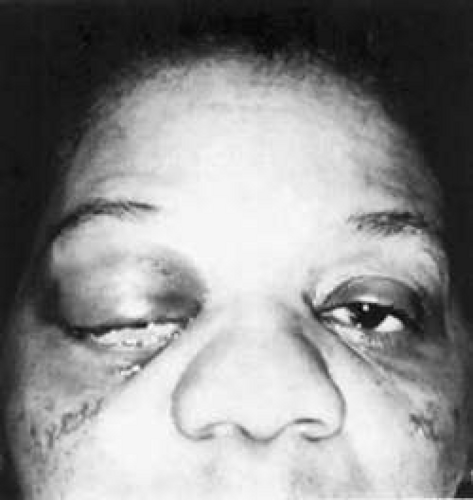 Fig. 35-2. A 54-year-old woman with acute orbital inflammation demonstrating significant proptosis and inflammatory signs involving the right orbit. In addition to severe pain, the patient had diplopia secondary to orbital edema and inflammation. |
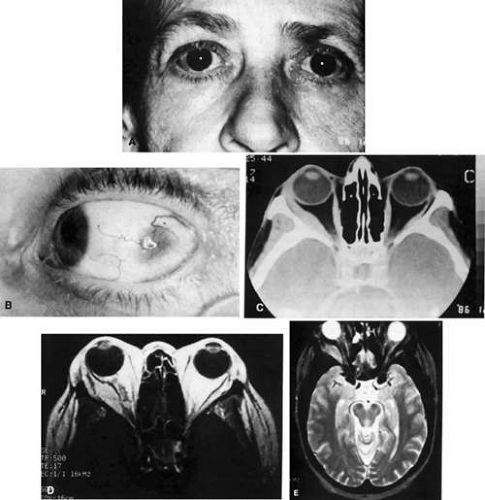 Fig. 35-3. A: A 62-year-old woman with diplopia and minimal right-sided proptosis. B: Demonstration of enlarged lateral rectus muscle visible subconjunctivally. C: Axial CT image confirming myositis involving lateral rectus muscle. Note that the muscle as well as its tendon are involved in the inflammatory process in contrast to muscle involvement in thyroid-related orbital disease. D: T1-weighted MRI image demonstrating myositis involving the lateral rectus muscle. In this case the muscle belly is enlarged, but the tendon appears relatively unaffected. E: T2-weighted MRI image of orbital myositis showing little difference in appearance of muscle between T1- and T2-weighted images. |
Acute Anterior Ocular-Orbital Inflammation.
Acute anterior ocular-orbital inflammation entity is characterized by inflammation of the globe, in particular, the sclera and the Tenon capsule, and the surrounding orbital tissues.3,4,6,28 The process may be either acute or subacute but is usually associated with decreased vision either due to uveitis, with or without an associated exudative retinal detachment, or papillitis and perineuritis.29 Chemosis, conjunctival injection, especially involving the episcleral vessels, eyelid edema, and acute pain radiating to the temple are also features of this process. Rarely there may be anterior segment cell and flare and even a sterile hypopyon. The differential diagnosis of anterior orbital inflammation includes orbital cellulitis, rupture of a dermoid cyst, a hemorrhagic episode associated with a lymphangioma, localized scleritis and uveitis, collagen vascular disease, rhabdomyosarcoma, or leukemic orbital infiltration.3,4
A type of uveal effusion syndrome may be present in association with anterior orbital inflammation.3,30,31 This often occurs in young men and is associated with exudative retinal detachment. These choroidal effusions have been reported to cause angle closure glaucoma due to anterior rotation of the ciliary body.32 Also present may be a diffuse and nodular choroidal swelling, thickened sclera, and retinal folds, in addition to the accumulation of fluid between the sclera and choroid. WG can be associated with similar findings.33,34 Hypertrophy and hyperplasia of the retinal pigment epithelium may also be present. This may mimic involvement of the choroid with melanoma, metastatic disease, or lymphoma.
Diffuse Idiopathic Orbital Inflammation.
The symptoms associated with diffuse idiopathic orbital inflammation are similar but more severe than those seen with anterior orbital inflammation.3,5,6 Visual loss due to papillitis and exudative retinal detachment, proptosis, and soft tissue inflammatory signs are present and are often more severe than in anterior orbital inflammation. In addition, it is associated with diplopia and limitation of extraocular motility. All tissues of the orbit are involved, including the fat, muscles, globe, and optic nerve. Rarely the inflammatory process may erode through bone and extend into the adjacent sinus cavities.4,11,12 Neoplastic processes, sarcoidosis, and orbital cellulitis are part of the differential diagnosis.
Focal Idiopathic Orbital Inflammation.
Localized inflammatory processes within the orbit often present as solitary neoplastic processes.3,5,6 These are characterized by proptosis with or without signs of acute inflammation. Diplopia may be present if there is also muscle involvement or due to surrounding edema. This process is often subacute in nature, as opposed to anterior orbital inflammation. Neoplastic disease again represents the most important diagnosis to exclude when considering these findings.
Orbital Apex and Superior Orbital Fissure Inflammatory Syndrome.
Idiopathic inflammation involving the orbital apex is associated with marked restriction of ocular motility as well as pain and minimal proptosis.13,35,36,37,38,39,40 The pain is present behind the globe and is boring and persistent in character. There is often little evidence of obvious inflammatory signs such as eyelid edema, chemosis, and conjunctival injection. Decreased vision and an afferent pupillary defect are frequently present due to either the presence of perineuritis or compression of the nerve by the surrounding edema. Computed tomographic (CT) evaluation of the orbit will demonstrate a diffuse inflammatory process in the posterior orbit with extension into the adjacent orbital fat. The inflammation may be noted to extend forward along the optic nerve and extraocular muscles or posteriorly through the superior orbital fissure.4,11
The Tolosa-Hunt syndrome is a specific form of apical idiopathic orbital inflammation involving the superior orbital fissure and the cavernous sinus.36,38,41,47 It is acute in onset and often progresses rapidly and can have a recurrent course.48 While usually unilateral, it may be bilateral. There is involvement of all the structures traveling through the fissure, including the third, fourth, and sixth cranial nerves and the first division of the trigeminal nerve, with hypoesthesia of the upper eyelid, forehead, and scalp as well as the optic nerve. Patients with this syndrome experience headache and ophthalmoplegia with pain described as constant, deep, and boring.38,49 Compromise of the superior orbital vein will result in edema of the eyelids and chemosis.46,50 The sympathetic innervation to the pupil may also be affected, resulting in pupillary dilatation and decreased pupillary motility. The inflammatory process appears to begin in the cavernous sinus and extend anteriorly. This accounts for the occasional involvement of the second division of the trigeminal nerve, which travels through the foramen rotundum and the inferior orbital fissure and the optic nerve, which is adjacent to the inflammed orbital apical structures. While generally diffuse in nature, this syndrome has been reported to occur in association with a discrete orbital mass.50
There are a number of diagnoses that must be considered in the evaluation of apical inflammation and the Tolosa-Hunt syndrome. These include trauma with or without hemorrhage, aneurysms and tumors within the cavernous sinus, parasellar neoplasms, metastatic disease, meningioma, and spread of adjacent sinus neoplasia or infection. Magnetic resonance imaging (MRI) of the cavernous sinus and, in some cases, orbital venography and fine needle biopsy of the cavernous sinus may be quite beneficial in evaluating the patient with symptoms associated with multiple cranial nerve palsies.35,50,51,52,53
Orbital Myositis
Orbital myositis (Fig. 35-3) is characterized by the sudden onset of painful ocular motility, diplopia, and conjunctival injection usually focally located over the involved muscle.6,9,54,55,56,57,58,59 There may also be eyelid edema and ptosis and proptosis.27,60,61 More than one muscle may be involved, although this is less common than single muscle involvement.62 The process may be recurrent despite adequate treatment.63 Inflammation of the trochlea is associated with pain on palpation in this area.64 Forced duction testing is usually positive. The most common muscles to be involved in this process are the superior and medial rectus muscles.
The most important differential diagnoses are thyroid-related orbitopathy and metastatic spread to an extraocular muscle. In both of these entities the extraocular muscles are involved only in the area of the muscle belly.65,66 In contrast, the entire muscle and tendon are usually involved in idiopathic orbital inflammation.65,67,68 There may also be some spread of the inflammatory process to the adjacent fat. In addition, myositis is almost always painful, while thyroid-related orbitopathy with extraocular muscle involvement is usually not associated with pain. Other signs such as eyelid retraction and lagophthalmos as well as abnormal thyroid function tests often accompany thyroid-related orbitopathy. Acquired retraction, however, can be the presenting sign of orbital myositis.69 Other diseases to be considered in the differential diagnosis include trichinosis, myasthenia gravis, and arteriovenous malformations. Trichinosis is usually associated with skin changes, myasthenia gravis is not associated with inflammation and responds to edrophonium, and arteriovenous malformations can usually be identified on computed tomographic evaluation when contrast is used. Orbital myositis has also been described following upper respiratory infections and in association with collagen vascular diseases, juvenile rheumatoid arthritis, Crohn disease, myocarditis, Kawasaki disease, and polyarteritis nodosa.55,62,70,71,72,73,74 A familial incidence has been reported, suggesting a potential genetic predispostion for orbital myositis.75
Orbital Angiitis.
Nonspecific inflammation of the orbital arteries and arterioles similar to that seen in connective tissue diseases but without any systemic associations has been termed orbital vasculitis.3,5,76 In this condition there is acute inflammation involving the small vessels of the orbit including capillaries, precapillary arterioles and post capillary venules, and adjacent connective tissue and muscle. There is usually perivascular hemorrhage due to vessel wall necrosis and destruction. The inflammatory infiltrate is polymorphic, consisting of lymphocytes, plasma cells, monocytes, and neutrophils. Patients present with nonspecific acute inflammatory symptoms and signs including pain, eyelid edema and ptosis, and chemosis and conjunctival injection. Proptosis is present in approximately 50% of patients. While the usual treatment of this condition is corticosteroids, resistant patients may require radiation, cyclophosphamide, or other chemotherapeutic agents.3
Orbital Thrombophlebitis.
Idiopathic inflammation of the orbital veins is associated with acute pain, chemosis, ocular motility disturbances, and decreased vision.4 These findings may be reminiscent of the Tolosa-Hunt syndrome.49,77 Thrombosis of the central retinal artery has been reported.78 Spontaneous orbital hemorrhage may occur, although no coagulation abnormalities are present.79 Although this condition is localized to the orbit, systemic associations have been described, including fatigue, cold extremities, gastrointestinal symptoms, vertigo, arthralgias, and memory impairment.80 The orbital pain experienced by these patients is unilateral and boring in character. It is worsened by eye movement and exposure to cold temperatures and is difficult to ameliorate with analgesics.80 Orbital phlebography has been used to study the venous abnormalities seen in this condition.49,81 This condition also may be associated with a variety of systemic diseases including typhus, parasitic infestations, mucormycosis, polyarteritis nodosa, and connective tissue diseases.
As with orbital angiitis, microscopic evaluation of tissue from orbital thrombophlebitis demonstrates an inflammatory reaction centered around blood vessels, in this case the orbital veins. This inflammation is segmental and may be associated with thrombosis. The walls of these veins may show evidence of infiltration with both neutrophils and lymphocytes without any granulomatous inflammation. As the inflammation subsides, fibrous tissue replaces the smooth muscle in the vessel walls.4 This fibrosis may extend into the surrounding orbital fat.
Dacryoadenitis.
In this conditions the lacrimal gland is enlarged and can be easily palpated in the superior lateral aspect of the orbit.3,5,9,82 It is usually tender and unilateral, although this is not always the case. The enlarged lacrimal gland will result in an S-shaped curve to the upper eyelid, and if the gland is large enough, the globe will be pushed down and medially as well as forward. Bilateral involvement should suggest the presence of a systemic disease such as sarcoidosis, lymphoma, Sjögren syndrome, malnutrition, metabolic derangement such as diabetes mellitus and cystic fibrosis, or drugs such as thiouracil. However, these diseases may also be associated with unilateral lacrimal gland enlargement. Viral and bacterial infections occasionally result in dacryoadenitis as well. Chronic lacrimal gland inflammation can also occur following radiation to the lateral orbit.
It is important to differentiate neoplastic processes involving the lacrimal gland from inflammation. On computed tomography, dacryoadenitis tends to show the gland to be enlarged in a oblong configuration from anterior to posterior as well as vertically with molding of the gland to the globe, inflammation of the adjacent tissue, and no erosion of the neighboring bony structures.3,4,9 Neoplastic processes are often more nodular and rounded and may be associated with bony erosion. However, biopsy of the gland is often needed to differentiate between the various causes of lacrimal gland enlargement.
Sclerosing IOID.
Sclerosing IOID appears to be a distinct entity from other IOIDs. It is characterized by a slow and relentless involvement of orbital structures. It is notoriously difficult to treat, with a poor response to steroids. Its pathogenesis is poorly understood but is thought to have an immunologic cell-mediated basis. It may represent a type of multifocal fibrosclerosis, which is a disease that includes retroperitoneal fibrosis, mediastinal fibrosis, Reidel sclerosing thyroiditis, and sclerosing cholangitis.83 Rootman et al., evaluated 16 cases of sclerosing IOID. They found an 11:5 male predominance with an age range of 8 to 81 years. The onset was usually unilateral with a chronic course. The clinical presentation could be divided into lacrimal and apical with signs and symptoms including infiltration (15/16), dull pain (13/16), mass effect (12/16), proptosis (11/16), mild inflammation (11/16), decreased extraocular motility (9/16), edema (9/16), diplopia (8/16), and vision loss (3/16). Histopathologically, there was a chronic paucicellular inflammatory infiltrate with desmoplastic stroma early in the disease. Treatment was inadequate, including corticosteroids (11/16), radiation for steroid failures (8/11), and observation (3/16). Complete resolution occurred in only 2/16 patients, with the rest having restricted motility (10/16) and blindness (3/16).84
Imaging Studies and Laboratory
The most useful imaging modality in the evaluation of the majority of orbital diseases is the CT scan (Figs. 35-1, 35-3). Advantages of the CT scan include cost, speed, wide availability, and good delineation of bony anatomy. At least two views of the orbit, most commonly axial and coronal views, with thin cuts should be ordered. Contrast improves the sensitivity and specificity of the study and should be ordered in most cases.85 In a series of 65 patients evaluated for IOID, orbital CT scan was the imaging study of choice in 91%.7 Common CT findings include retrobulbar fat infiltration and apical fat edema, proptosis, enlargement of extraocular muscles with or without tendon involvement, uveal scleral thickening, Tenon capsule edema, and lacrimal gland enlargement or infiltration.7 In addition, it will show if there is any associated sinus disease or bony erosion, which would suggest a neoplastic process or one of the necrotizing vasculitides such as WG.
MRI is useful if more definition of soft tissue is required. Any process with suspected intracranial spread should also be evaluated with MRI because of its superior sensitivity for brain pathology. Disadvantages of MRI are increased cost, length of exam, decreased availability, contraindication in patients with ferromagnetic implants, and potential for inducing claustrophobia.85 Contrast enhanced MRI with axial and coronal views and fat suppression should be ordered.86 IOID is typically revealed as a lesion hypointense to fat on T1-weighted images and isointense or minimally hyperintense to fat on T2-weighted images.87
Other imaging studies that may be useful include ultrasonography, positron emission tomography (PET), orbital venography, and gallium scanning. Ultrasonography is helpful for demonstrating exudative retinal detachment, edema in Tenon fascia, enlargement and “doubling” of the optic nerve shadow (the T sign), enlargement of the extraocular muscles, and the presence of a distinct orbital mass.4,77,81,82 Orbital venography may be helpful in demonstrating vascular deformities that are seen in the Tolosa-Hunt syndrome and occasionally in other forms of inflammatory orbital inflammation.58 Gallium scanning has also been used to image in order to differentiate it from other entities involving the orbit.88
Patients suspected of IOID should have a full hematologic work-up with complete blood count, electrolytes, erythrocyte sedimentation rate (ESR), antineutrophil cytoplasmic antibodies (ANCA), antinuclear antibodies, anti-dsDNA, angiotensin converting enzyme (ACE), rapid plasma reagin test, serum protein electrophoresis (SPEP),86 and cerebral spinal fluid (CSF) analysis. The ESR may be elevated in cases of orbital inflammation, but this should bring to mind the possibility of a concurrent systemic disease such as one of the collagen vascular diseases.3,53,89 The antinuclear and anti-dsDNA antibodies may also be positive in these diseases. Testing for ANCAs, which are highly specific for WG, will also help to differentiate this entity from idiopathic causes of scleritis and idiopathic orbital inflammation.90 Elevation of the ACE level, which occurs in granulomatous diseases, is suggestive of sarcoidosis. Cerebrospinal fluid evaluation is rarely helpful, but pleocytosis has been reported due to meningeal irritation.3 Also, CSF can be useful in questionable cases to rule out lymphoma.86
One of the most important diagnostic aids in the evaluation of idiopathic orbital inflammation is the utilization of a trial of high-dose corticosteroids,5,9,91 as the disease is almost always exquisitely responsive to systemic steroids. An 80-mg daily oral dose for a week will usually result in significant improvement or complete resolution of the inflammatory process. If this does not occur, consideration must be given to other diagnoses, and orbital biopsy should be performed in order to obtain a tissue diagnosis.5,51,91,92
Histopathology
In general idiopathic orbital inflammation (Figs. 35-5, 35-6) is polymorphic in nature.3,4,9,24,91,93,94 The cellular response seen in this disease consists of lymphocytes, plasma cells, macrophages, histiocytes, occasional neutrophils and eosinophils, epithelioid cells, and fibroblasts. These are present in varying numbers depending upon the chronicity of the inflammatory process. Intranuclear Dutcher bodies and intracytoplasmic Russell bodies may be present within many of the plasma cells, as they actively participate in the inflammatory response. Children tend to have a significant number of eosinophils in their biopsy specimen. Lipogranulomatous inflammation with fat necrosis may also be present. However, the presence of noncaseating granulomas is relatively uncommon.95 This is often associated with foreign body giant cells that are responding to the release of lipid. Perivascular lymphocytic cuffing and capillary proliferation are quite common.4 Occasionally, lymphoid follicles may also be seen.
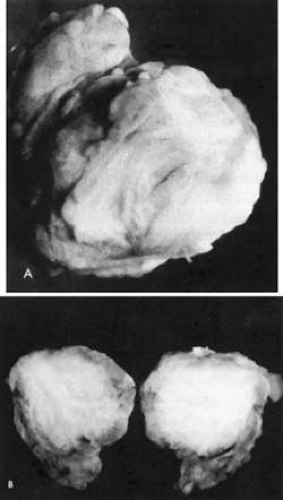 Fig. 35-4. Gross specimen of old, burned-out, involuted granuloma from a patient with idiopathic orbital inflammation. Note lack of vascularity. |
 Fig. 35-5. Section from globe removed (in era before corticosteroid therapy) because the inflammatory process had progressed inexorably to dense scar formation with resultant painful eye. Note that the dense ligneous tissue above globe has fused with sclera (hematoxylin-eosin, 4X). |
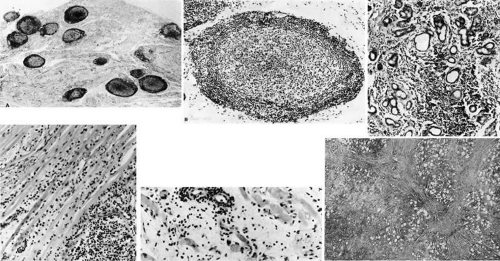 Fig. 35-6. Histopathologic sections demonstrating the various pathologic findings associated with idiopathic orbital inflammation. Most commonly these include lymphoid follicles, granulomas, collagen deposition, and a diffuse mixed inflammatory cell infiltrate. The normal tissue architecture is frequently disrupted by these changes. A: Many lymphoid follicles are scattered throughout orbital tissue (hematoxylin-eosin, 63X). B: Cells comprising the follicular center are lighter and larger than mantle of mature lymphocytes that surround the germinal zone (hematoxylin-eosin, 160X). C: Lacrimal gland elements have undergone atrophy in an advanced example of idiopathic dacryoadenitis. Fibrosis and lymphocytes have replaced a considerable amount of gland parenchyma (hematoxylin-eosin,94X). D: Myositis in which lymphocytes are loosely aggregated below the center and infiltrate between extraocular muscle fibers (hematoxylin-eosin, 160X). E: Cuffing of small vessels by mature lymphocytes. Note the loose edematous interstitium between disrupted muscle fibers (hematoxylin-eosin, 240X). F: Progressive fibrosis of retrobulbar fat (hematoxylin-eosin, 25X). |
Vasculitis is a very unusual feature of idiopathic orbital inflammation.4,42 When this is present there is destruction of the muscularis layer of the orbital vessels by lymphocytes and neutrophils, as well as eosinophilic infiltration. There is also focal vascular necrosis with leukocytoclasis or collections of debris from degenerated neutrophils.4,5 When it appears that a vasculitis is present, consideration must be given to a diagnosis of polyarteritis nodosa, WG, or one of the collagen vascular diseases.
With progression of the disease process, there is increased fibrosis and collagen deposition, which separates the various inflammatory elements. There is also an associated thickening of the connective tissue within the extraocular muscles. With chronicity there will be actual loss of muscle tissue and its replacement with connective tissue. In the lacrimal gland there is hyperplasia of the periductal and periacinar connective tissue. As this progresses, there will be actual destruction of the lacrimal acini.82 In the sclerosing types of idiopathic orbital inflammation, there is extensive fibrosis surrounding and replacing all normal orbital tissues.82,96,97 There is only a minimal cellular response present in such instances. At times this connective tissue reaction may be so dense as to have the consistency of cartilage.
It is important to differentiate between idiopathic orbital inflammation and lymphoma or its precursors.98 Lymphoid follicles, a polymorphic infiltrate, and the absence of monoclonality are essential aspects of idiopathic orbital inflammation.9 By contrast, lymphomas and atypical lymphoid hyperplasia tend to be monomorphic in nature, without lymphoid follicle, and monoclonal. Capillary proliferation is not common in lymphomas, nor is the presence of histiocytes and macrophages.
Treatment
The mainstay of treatment for IOID is centered around anti-inflammatory medication. It may be possible to treat some forms of orbital inflammation with nonsteroidal anti-inflammatory medications such as indomethacin.99 However, this type of therapy has not been well studied in the management of this disease process. In general, idiopathic inflammation of the orbit is quite sensitive to corticosteroids.4,5,9,58,91,100 Prednisone in a dose of 80 to 100 mg orally will usually result in a marked improvement in symptomatology within 2 to 3 days after their institution. In fact, this is one method for making the diagnosis of this disease. Steroids should be continued for 2 to 3 weeks and then slowly tapered over an additional 3 weeks. Failure to do this will result in recurrence of the orbital inflammation. Given the dramatic response to oral prednisone, intravenous steroids should be reserved for patients with rapidly progressing symptoms, vision loss, or refractory disease. There have been several reports of pulse intravenous methylprednisolone ranging from 1 mg/kg per day to 1,000 mg total dose per day for aggressive IOID.86 It is important to determine prior to beginning steroid treatment if a patient has ulcers involving the digestive tract; any systemic infectious processes, especially herpes virus, HIV, fungal disease, tuberculosis, and amebiasis; recent myocardial infarction; or diabetes mellitus, as the use of steroids in these instances may result in significant exacerbation of the pre-existing systemic disease.
The current belief is that in the majority of cases corticosteroids are curative, with only some patients having recurrence of their disease process or failing to adequately respond. Recent evidence suggests that the cure rate may be lower than previously thought. Cure rates of 37% to 63% and recurrence rates of 21% to 52% have been reported.7,101 These series were from major referral centers and may represent a higher rate of steroid failure than in the general population. In patients who experience a recurrence, a second course of steroids can be instituted over a longer period of time and with a much-prolonged taper. In patients with an inadequate response, immunosuppressive therapy or radiotherapy can be considered after a tissue diagnosis is obtained.3,5,91,102 The greater the amount of fibrosis that is present within the orbit, the less responsive the inflammatory process is to steroid administration.5,58,96,103 It is important to biopsy all apparently inflammatory orbital lesions that fail to respond adequately to a course of steroids in order to eliminate other lesions that may appear to be inflammatory in nature.5,91
When radiotherapy is administered to lesions that have failed steroid treatment, the usual dose is 10 to 30 Gy given in divided doses over a 2- to 3-week period.104,105,106 This is similar to the dosage used in lymphoid tumors, and it may be that a refractory orbital inflammatory lesion is in reality a misdiagnosed lymphoid tumor.4 Rates of improvement have been shown to be 72% to 84% with these doses.98,99,100,107,108 The advent of new radiation treatment modalities such as radiosurgical techniques like cyberknife or gamma knife have greatly improved the safety and utility of radiation. Radiosurgery techniques increase the conformality to the planned treatment zone, allowing for greater precision while limiting the damage to surrounding normal tissue.109 In rare instances, as in sclerosing IOID, there may be a less than optimum response even to radiation.
In refractory cases or in patients who cannot tolerate systemic steroids, adjuvant treatment can be tried. The spectrum of these types of treatments are wide and still evolving. Options include surgical debulking, intralesional steroid injection, cytotoxic agents, immunosuppressants, plasmapheresis, and biologic treatments or immunomodulators. Surgical biopsy or debulking is useful in ruling out other causes such as lymphoma, WG, or metastasis. It is important to consider fibrous histiocytoma as a possible diagnosis in very refractory cases of sclerosing “pseudotumor”.91,110 The cytotoxic agent cyclophosphamide and immunosuppressants such as methotrexate and cyclosporin have all been reported to be effective.111,112 The newest class of adjuvant treatment, called biologic treatment, involves monoclonal antibodies that act as immunomodulators. One group of these antibodies works to inhibit TNF-α, its receptor, or both. There are three TNF-α blockers with FDA approval: infliximab, etanercept, and adalimumab.113 Infliximab and adalimumab have been successful in treating recalcitrant IOID.114,115,116 Another immunomodulator is rituximab. This compound is a genetically engineered monoclonal antibody directed against the CD20 antigen found on all B cells. It is thus capable of inciting a cytotoxic effect on CD20-positive cells via antibody-dependent cell-mediated and complement-dependent mechanisms. This drug has been used successfully in the treatment of CD20-positive tumors such as nonHodgkin lymphoma of the orbit.117,118 The authors have used rituximab in combination with cyberknife radiosurgery to successfully treat sclerosing IOID.
It is extremely unusual for a patient to be totally unresponsive to all treatment modalities. However, if there is persistent debilitating pain and loss of vision, orbital exenteration can be carried out as a last resort.119 Trigeminal ganglion blocks with glycerin can also be tried for pain control, although this should only be performed by a physician experience in this procedure.
OTHER LOCALIZED IDIOPATHIC ORBITAL INFLAMMATORY PROCESSES
Nodular Fasciitis
Nodular or pseudosarcomatous fasciitis (Fig. 35-7), while not usually considered part of the spectrum of idiopathic orbital inflammation, does represent a localized inflammatory disease of idiopathic origin. It appears to be a reactive process involving a proliferation of immature connective tissue elements along with acute and chronic inflammatory foci.120,121,122,123,124 This entity can develop periorbitally in the eyelid as well as in the epibulbar tissue. It may also occur in the anterior or deep orbit. There have been 23 cases of nodular fasciitis reported in the English language literature. The majority of these patients were adults, but 8 of 23 of these patients were younger than 20 years old, one of which was an infant.125 It appears as a rapidly growing mass arising over several weeks or months. It may or may not be associated with pain. On imaging studies, it is difficult to distinguish nodular fasciitis from malignant soft tissue tumors such as rhabdomyosarcoma and granulocytic sarcoma. For this reason, surgical excision is recommended.125 At surgery, the lesion appears fairly well demarcated and slightly reddish in color.
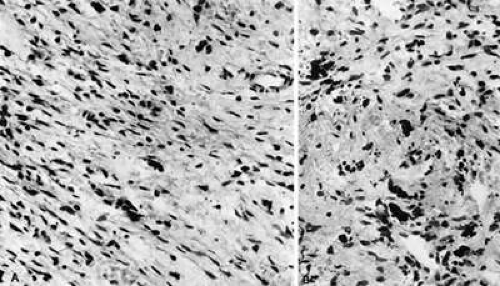 Fig. 35-7. A: Fasciitis. “Pseudosarcomatous” reactive inflammatory condition is composed predominantly of a loose arrangement of fibroblasts with scattered chronic inflammatory cells (hematoxylin-eosin, 240X). B: Fasciitis. Conspicuous hemosiderin-laden macrophages are seen in the center of this photomicrograph. The presence of abundant numbers of capillaries in the inflammatory tissue results in hemorrhage and the presence of blood breakdown products (hematoxylin-eosin, 240X). |
Histopathology.
Microscopically there is a proliferation of immature and active-appearing fibroblasts within the orbital fascial planes.120,121,122,123,124 These fibroblasts may be loosely connected with occasional myxoid foci interposed. Slits often appear between the fibroblasts, which appear to be quite plump and have basophilic cytoplasm. There may be rare mitoses among these cells. There is only a minimal amount of collagen deposition present. The appearance of a gradient from immature fibroblastic elements to more mature fibroblasts is characteristic of this lesion. Also associated with this process are acute and chronic inflammatory cells, capillary and endothelial cell proliferation, and occasional giant cells. Occasionally elements of this inflammatory process can be seen to invade surrounding muscle fibers and orbital fat. The proliferating capillaries are friable and bleed. When this occurs, hemosiderin-laden macrophages can be identified in the tissue. Electron microscopic evaluation of the cells present in this process shows that most are myofibroblasts. These are cells containing rough endoplasmic reticulum and no basement membrane like fibroblasts but also have cytoplasmic actin filaments with fusiform densities like smooth muscle cells.4
Treatment.
While this process often appears to be malignant, especially with its rapid growth and the immature appearance of the fibroblasts that are present, it is a benign inflammatory condition. Fibrosarcoma does not usually have an significant inflammatory component nor does it demonstrate both immature and mature tissue elements. Rhabdomyosarcoma is a consideration when this lesion is present in a child. Myositis is also a possible member of the differential diagnosis, although this entity is more confined to the muscle tissue than the fascial planes. All of the reported cases of orbital nodular fasciitis were treated with surgical excision. Given its well-circumscribed nature, complete surgical excision rather than a partial diagnostic biopsy should be performed. This usually proves to be curative.125
Benign Lymphoepithelial Lesion
Lacrimal gland enlargement in the absence of systemic disease without dry eye symptomatology has been termed benign lymphoepithelial lesion (BLL) (Fig. 35-8).4,126 Previously this entity was known as Mikulicz syndrome or disease. However, due to confusion as to the exact definition of these terms, they have been abandoned. Histologically, sheets of lymphocytes and plasma cells, which replace the normal acinar and ductular structure of the gland, characterize this condition. Lymphoid follicles may be present. The remaining ductular elements may proliferate to form solid nests of cells outlined by excessive basement membrane production. Hyalinization may sometimes occur within these ductular remains. The cells within these nests are for the most part epithelial elements of the ducts. These structures are commonly called myoepithelial islands due to the incorrect belief that normal ducts have myoepithelial cells present in their wall. This has been disproved on electron microscopic evaluation.127 On superficial examination these formations may resemble lymphoid follicles. The presence of these ductular nests helps to differentiate this disorder from lymphomatous involvement of the gland, which is not associated this these structures. From one third to one half of patients with Sjögren syndrome may have similar nests of ductular elements. Historically, this has led to some confusion as to whether BLL was a distinct entity from Sjögren. This confusion has been resolved by a study that demonstrated that BLL has higher serologic levels of IgG4 with infiltration of the lacrimal glands with IgG4-positive plasmocytes.128 Another study showed that the acinar cells in BLL did not undergo apoptosis as in Sjögren, and therefore, sicca syndrome is not observed in BLL.129 The difference between BLL and IOID involving the lacrimal gland is based on the more monomorphic infiltrate seen in BLL in contrast to the relatively more polymorphic infiltrate seen in IOID. In addition, IOID contains more plasma cells and fibrotic tissue and is associated with lymphoid follicles with active germinal centers. Finally, the lacrimal gland capsule is usually preserved in BLL, in contrast to IOID, which is a more destructive process.
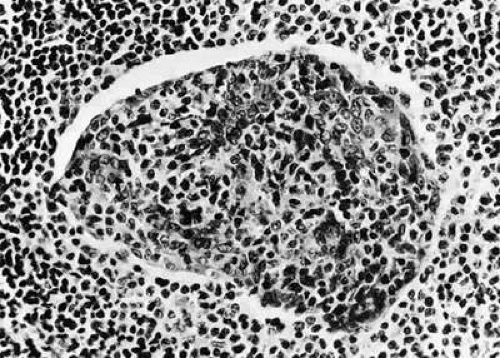 Fig. 35-8. This epimyoepithelial island is composed of cytologically benign cells, which have retracted from surrounding inflammatory infiltrate. The island is indicative of a inflammatory disorder and weighs heavily against interpreting lymphocytic infiltrate as lymphoma (hematoxylin-eosin, 76% of 320X). |
REACTIVE ORBITAL INFLAMMATION
Amyloidosis
Amyloidosis (Fig. 35-9) is a disease process characterized by the deposition of amorphous hyaline material in various body tissues including nerves, blood vessels, muscles, skin, spleen, adrenal glands, liver, heart, tongue, and orbit.130 Two types of primary amyloidosis have been identified. One is a localized process restricted to the ocular and orbital structures without systemic manifestations.4,131,138 Radiologically, this type has a homogeneous mass with irregularity of adjacent bone and intralesional calcification on CT scan.137 The second is characterized by ocular as well as systemic deposition of amyloid.4,139 This may also occur in a familial form, with amyloid infiltrating the heart, peripheral nerves, and blood vessels.4 Retinal deposits occur due to extension of the amyloid from the retinal vasculature, and infiltration of the trabecular meshwork can result in glaucoma. Secondary amyloidosis occurs as part of a variety of chronic disease entities including tuberculosis, rheumatoid arthritis, leprosy, osteomyelitis, multiple myeloma and other plasma cell dyscrasias, and occasionally lymphoma.4 A systemic evaluation for these conditions should be performed in any patient presenting with amyloidosis.
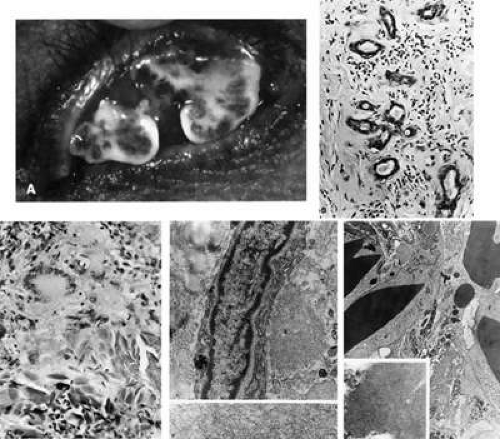 Fig. 35-9. A: Amyloidosis. Recurrent conjunctival vegetations with mild chronic inflammation and tremendous amyloid content. B: Amorphous intercellular material has replaced most of the lacrimal gland, with few glandular units surviving. There is an infiltrate of chronic inflammatory cells, many of which are plasma cells (hematoxylin-eosin, 74% of 240X). C: Amorphous eosinophilic material, giant cell, epithelioid cells, and fragments of needlelike particles, which have elicited a foreign-body reaction, are shown. The condition is termed tissue proteinosis (hematoxylin-eosin, 240X). D: This electron micrograph from a case of lacrimal gland amyloidosis shows a surviving cell, its identity unascertainable, with cytoplasmic filaments and profiles of rough endoplasmic reticulum, surrounded on the right by amyloid fibrils and on the left by collagen. There is an osmophilic inclusion at the bottom left of the cell. In the lower panel, amyloid material is seen as slender (100-Å wide) nonbranching fibrils (top, 8,000X; bottom, 25,000X). E: Osmophilic material with crystalline substructure (shown in inset) is being engulfed by degenerating giant cells and macrophages, which have abundant cytoplasmic filaments (F). It is possible that intracytoplasmic filaments, many of which may have been extended into the intercellular space after dissolution of cell membranes, are responsible for borderline amyloid stain. Latticelike material is proteinaceous, with resemblance to crystallized immunoglobulin. (5,000X; inset 60,000X) |
Symptomology and clinical findings associated with this disease process depends upon the tissues that are invaded by the amyloid. Most lesions hemorrhage quite easily due to vascular infiltration. Patients can present with periorbital or subconjunctival hemorrhage. Loss of extraocular motility and ptosis from muscle involvement, vitreous opacities and retinal deposits secondary to intraocular infiltration, and pupillary abnormalities from deposits in the cervical lymph nodes can occur.131,140,141,142 Ocular involvement in secondary amyloidosis is unusual. However, the presence of waxy yellow skin nodules that bleed with only slight trauma is characteristic of systemic amyloidosis. Congestive heart failure, gastrointestinal motility disturbances and malabsorption, hepatosplenomegaly, and proteinuria and the nephrotic syndrome occur with systemic involvement of the associated organ systems.
Localized amyloid deposits in ocular disease can occur in any portion of the orbit and globe. Systemic disease is not usually associated with conjunctival, lacrimal gland, or orbital amyloid deposition.131,133,141,143 This is in contrast to skin deposition, which is usually associated with systemic amyloid. Pseudoptosis may be present due to a mass effect, although deposition in the Müller muscle and the levator aponeurosis is also a cause of ptosis.134,140 Deposits palpated through the eyelid feel somewhat cystic in contrast to the more superficial infiltrates that appear as friable excrescences or nodules protruding from the conjunctiva. Lacrimal gland enlargement and proptosis can also occur due to localized amyloid deposition.131 Orbital and extraocular amyloid is not associated with pain. Vision is only rarely affected, usually from optic nerve infiltration.144 Double vision may occur when there is amyloid deposition within the extraocular muscles.142,145
Histopathology.
Amyloid deposits appear as paucicellular hyalinized amorphous material. A surrounding foreign body–type granulomatous response is present in reaction to this material.4,146 Scattered elongated fibroblasts and plasma cells are also present throughout the infiltrating material. Perivascular infiltration is characteristic of this process. Amyloid deposits are eosinophilic on routine hematoxylin and eosin staining, appearing similar to extensive collagen deposition. These deposits also stain positively with PAS, Congo red, crystal violet, where the exhibit metachromasia, and thioflavin T stain. The material is birefringent and dichroic when viewed by polarization microscopy.
When amyloid is examined by electron microscopy, nonbranching thin fibrils with a distinct periodicity are seen.130 Antibodies against immunoglobulin fragments may react with localized amyloid deposits. Monoclonal staining of lymphocytes and plasma cells seen in association with amyloid deposits may occur.147
A wide variety of proteins are present in amyloid deposits. These proteins differ between individuals presenting with this disorder and between different forms of the disease. It appears that a circulating protein is converted when phagocytized into fibrils that, when coalesced, form amyloid deposits.146,148,149 Protein AL is present in patients with multiple myeloma and in primary systemic amyloidosis.146 This protein consists of complete kappa or lambda chains with or without N-terminal fragments from these light chains. Bence Jones protein from multiple myeloma patients may be modified to from protein AL. Protein AA is part of amyloid deposits associated with inflammation.150 These protein may be a breakdown product of protein SAA that is produced by hepatocytes. Protein SAA is an acute phase reactant that becomes markedly elevated in chronic infections and idiopathic inflammation. Other proteins that have been isolated in cases of amyloid include protein AP, normally found in plasma, protein AFp, related to prealbumin, and protein AEt, related to calcitonin.4,148
Giant Cell Reparative Granuloma
Giant Cell Reparative Granuloma is an idiopathic inflammatory intraosseous tumor that rarely occurs in the orbit.151,152,153 Usually it involves the maxilla and orbital floor and the mandible. Young persons are most often involved, but the condition has been reported in a patient as old as 85.154 Trauma is frequently associated with the occurrence of this tumor, which is characterized by the presence of multinucleated giant cells within a stromal matrix of spindle cells. In addition to inflammatory cells, there are hemorrhage and vascular proliferation within the tumor. Giant cell reparative granuloma has been associated with cherubism, an autosomal dominant disease characterized by painless symmetrical swelling and polycystic destruction of the mandibular bone.155,156 The differential diagnosis of this lesion includes the brown tumor of hypoparathyroidism, true giant cell tumors, malignant giant cell soft tissue tumors, and aneurysmal bone cysts. In brown tumor there is a hyperfunctioning parathyroid gland with increased calcium and parathormone levels.157,158 This tumor may also occur in association with secondary hyperparathyroidism from metabolic derangement, as is seen in chronic renal failure. In true giant cell tumors there is an even distribution of these multinucleated cells throughout the surrounding spindle cells.4 This contrasts to the zonal arrangement of giant cells found in giant cell reparative granuloma and brown tumor. In these zonal areas, giant cells surround foci of hemorrhage and inflammation. Malignant giant cell tumor of soft parts is multinodular and contains very large multinucleated giant cells with stromal anaplasia, areas of fibrosarcoma, and osteoblastic transformation.159 Finally, aneurysmal bone cysts have giant cells and reactive bone formation similar to brown tumor and other giant cell tumors.4 However, the characteristic feature of this entity is the presence of a nonendothelial-lined cavity. The treatment of all giant cell tumors involving bone is curettage and excision of all abnormal bony elements.
Other Reactive Orbital Inflammations
Retained orbital foreign bodies, especially vegetable matter and other organic substances, can result in chronic orbital inflammation including pain, proptosis, chemosis, and periorbital edema.160,161 Chronic discharge, erythema, and fistulous tracts can also occur. Foreign body granuloma formation as well as areas of acute inflammation are characteristic of this process. CT and MRI are helpful in locating the offending foreign body.
Ruptured dermoid and hematic cysts, orbital hemorrhage, and cholesterol granulomas can also elicit an acute granulomatous inflammatory process.160,162,163,164,165,166 These ruptures usually occur secondary to trauma, although hematic cysts may rupture spontaneously. Ruptured dermoid cysts show cholesterol crystals and cyst wall remnants surrounded by the granulomatous inflammatory response. Ruptured hematic cysts are associated with hemosiderin deposition, macrophages, and fibrous capsule formation around blood breakdown products.
Pain, proptosis, periorbital edema and erythema, chemosis, and extraocular motility disturbances can occur as an inflammatory reaction to acute sinusitis.167,168,169,170 Patients will also manifest symptoms of sinusitis including sinus pain and congestion of the nasal passages, as well as systemic symptoms of fever, malaise, and distress. Orbital infection is not present. Obstruction of the venous outflow from the orbit and sinuses adds further to the increasing orbital edema. In severe cases, visual loss from optic nerve compression or corneal drying and exposure can occur. CT of the orbits and sinus cavities is an essential part of the evaluation of this complex of symptoms in order to eliminate other causes of acute orbital inflammation. Successful treatment of this condition is predicated upon aggressive antibiotic and surgical management of the underlying sinus infection.
ORBITAL INFLAMMATION ASSOCIATED WITH SYSTEMIC DISEASE AND VASCULITIS
Wegener Granulomatosis
Wegener granulomatosis (WG) (Figs. 35-10, 35-11) is characterized by inflammatory lesions of the upper respiratory tract, lower respiratory tract, and kidneys and, to varying degrees, by generalized small vessel vasculitis of other structures, including the eye. A limited form of the disease has been described that spares the kidneys, frequently involves the head and neck region, and may not progress to the systemic form.171 The definitive diagnosis is confirmed by biopsy.
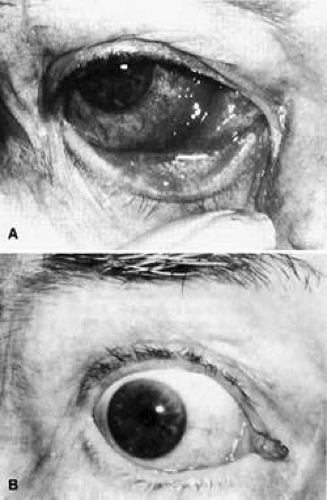 Fig. 35-10. A: This 68-year-old woman presented with acute orbital inflammation, pain, and diplopia. Clinical evaluation demonstrated a medial orbital mass. Biopsy of the orbital mass demonstrated significant vasculitic and granulomatous changes felt to be consistent with Wegener granulomatosis. No systemic disease was present. B: Appearance of same patient after treatment with prednisone and cyclophosphamide, demonstrating complete resolution of the inflammatory process. |
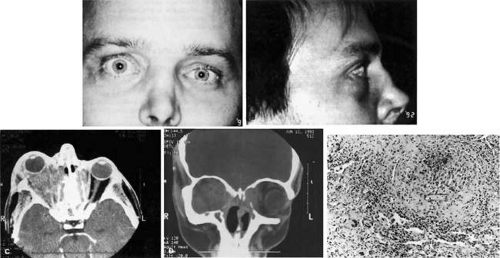 Fig. 35-11. A: This 31-year-old man has had Wegener granulomatosis for 2 years. His disease process is stable on chronic corticosteroid therapy. He has no evidence of systemic disease. Note right-sided proptosis and hyperglobus. B: Profile of same patient demonstrating collapse of nasal bridge from bony destruction secondary to Wegener granulomatosis. Note presence of swelling in lower eyelid. C: Axial CT image from the same patient demonstrating significant bilateral disease and bony destruction. Despite the extent of the orbital process on the right, the patient does not have diplopia. D: Coronal CT image showing destruction of medial orbital walls, vomer, and orbital septum. E: Wegener granulomatosis. Pulmonary biopsy specimen from patient with orbital signs contains an almost obliterated vessel to right of center and scattered giant cells on left (hematoxylin-eosin, 160X) |
As described below, ocular manifestations include orbital inflammation, scleritis, keratitis, and uveitis. The ocular involvement can occur from extension of sinus and nasal lesions or from focal small vessel vasculitis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


