New Pharmacotherapies for Age-Related Macular Degeneration
Scott D. Schoenberger
Paul Sternberg Jr.
INTRODUCTION
The pathogenesis of age-related macular degeneration (AMD) is highly complex and incompletely understood. Its causes are multifactorial, with genetic predisposition and environmental factors likely playing a role. Numerous molecular and biochemical pathways have been suggested to be involved, leading to optimism for several potential therapies targeting these pathways. Perhaps the greatest success has been in the treatment of exudative AMD, with the emergence of several efficacious agents directed against vascular endothelial growth factor (VEGF). However, the only current therapy with documented benefit for dry AMD has been the use of nutritional supplements, as demonstrated in the Age-Related Eye Disease Study. A wide variety of therapies directed toward different pathways are being investigated in preclinical testing and human clinical trials for both forms of AMD.
Chronic inflammation, complement deposition, oxidative stress, angiogenesis, and visual cycle toxic by-products have been associated with the development and progression of AMD (1,2,3,4,5). In dry AMD, inflammatory cells, complement components, amyloid-β (Aβ), and oxidized lipids have been shown to localize within or near drusen (4,5,6,7). In addition, chronic inflammatory cells, complement components, and cytokines increase choroidal neovascularization (CNV) formation or are overexpressed in CNV (8,9). Laboratory in vitro studies and animal research have shown that these pathways may be interconnected. Interferon-gamma (IFN-γ) is a proinflammatory cytokine that induces complement factor H (CFH) and promotes VEGF secretion (10,11). Aβ colocalizes with activation-specific fragments of complement C3 and activates the complement cascade (12). Oxidative stress generates reactive oxygen species (ROS) within lipofuscin, and this exposure leads to the deposition of complement components, microglia, and other inflammatory cells in the retina (3,13).
The above pathways have been targeted in animal studies and preclinical studies, and many are currently undergoing clinical testing in human patients with AMD.
INFLAMMATION
Chronic inflammation has been associated with multiple neurodegenerative disorders of aging (14), including AMD (4). Autoimmunity has been linked with AMD, as a majority of patients in one study had elevated serum retinal autoantibodies, as compared to 9% in controls (15). Serum C-reactive protein (CRP) levels have been shown to be elevated in some studies of patients with AMD (16). On a histologic level, several studies have linked AMD with inflammation. Inflammatory cells and proinflammatory cytokines have been demonstrated to contribute to both dry and exudative AMD. Macrophages and giant cells localize near drusen, at CNV, and at breaks in Bruch’s membrane (8). Macrophages
induce proliferation and migration of vascular endothelium, increasing CNV (17). Activated microglia, as part of the innate immune system, migrate within the retina, ingest debris, and produce cytokines (18). Inflammatory cytokines including tumor necrosis factor-alpha (TNF-α) and interleukin-1 (IL-1) have been shown to promote intercellular adhesion molecule-1 (ICAM-1) formation in the retinal pigment epithelium (RPE) and vascular endothelium, leading to a further increase in inflammatory cells, creating a self-perpetuating cycle of inflammation. IFN-γ is an important modulator of the immune system, up-regulating complement components and other cytokines, leading to increased leukocyte activation (11). IFN-γ transgenic mice developed inflammatory cell infiltration and photoreceptor loss (19). Thus, targeting chronic inflammation may be beneficial for both atrophic and exudative AMD.
induce proliferation and migration of vascular endothelium, increasing CNV (17). Activated microglia, as part of the innate immune system, migrate within the retina, ingest debris, and produce cytokines (18). Inflammatory cytokines including tumor necrosis factor-alpha (TNF-α) and interleukin-1 (IL-1) have been shown to promote intercellular adhesion molecule-1 (ICAM-1) formation in the retinal pigment epithelium (RPE) and vascular endothelium, leading to a further increase in inflammatory cells, creating a self-perpetuating cycle of inflammation. IFN-γ is an important modulator of the immune system, up-regulating complement components and other cytokines, leading to increased leukocyte activation (11). IFN-γ transgenic mice developed inflammatory cell infiltration and photoreceptor loss (19). Thus, targeting chronic inflammation may be beneficial for both atrophic and exudative AMD.
Nonsteroidal Anti-Inflammatory Drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) are used systemically for their anti-inflammatory, analgesic, and antipyretic properties. They are used topically for inflammation, allergic conjunctivitis and keratitis, inhibition of miosis, and cystoid macular edema (20). Newer formulations have better penetration into the posterior segment of the eye. NSAIDs inhibit the cyclooxygenase (COX) pathway, including the COX-1 and COX-2 enzymes. COX-2 has been detected in the RPE and vascular endothelium within CNV (21). It is an inducible enzyme that produces prostaglandins that increase vascular permeability and modulate the expression of VEGF and its receptors (20).
Topical NSAIDs have been investigated in exudative AMD. Zweifel et al. (22) investigated topical bromfenac as an adjunct to intravitreal anti-VEGF therapy in patients with persistent subretinal or intraretinal fluid. They found no statistically significant benefit over 2 months. Chen et al. (23) similarly found no statistically significant benefit to the addition of topical nepafenac to intravitreal anti-VEGF agents in patients with persistently active exudative AMD. A prospective, randomized study found the addition of topical diclofenac to photodynamic therapy (PDT) to be no superior to PDT alone (24).
Intravitreal NSAIDs have been tested in animal models (25). Studies have shown intravitreal NSAIDs to have powerful anti-inflammatory effects in rabbit models (26). In rats, intravitreal ketorolac reduced laser-induced CNV leakage and vascular budding (25). Intravitreal NSAIDs have been evaluated more commonly in humans with diabetic macular edema and uveitis, but in one report of intravitreal diclofenac for various etiologies, increased visual acuity (VA) was noted in both patients with exudative AMD (27).
Corticosteroids
Corticosteroids have several anti-inflammatory effects: they induce lipocortin synthesis, which inhibits phospholipase A2 and arachadonic acid formation, leading to a decrease in prostaglandins and leukotrienes through the COX and lipo-oxygenase pathways (28); they inhibit the production of IL-1 and TNF-α (4) and down-regulate cyto-kine-induced expression of adhesion molecules, further inhibiting adhesion and migration of inflammatory cells (28); within the eye, they reduce vascular permeability, stabilize the blood-ocular barrier, and suppress VEGF expression (29).
Dexamethasone and triamcinolone have been studied in clinical settings. Intravitreal dexamethasone has been combined with anti-VEGF therapy and PDT. Intravitreal triamcinolone acetonide has been evaluated in combination with anti-VEGF and/or PDT, with mixed results (28). The addition of intravitreal dexamethasone or triamcinolone may decrease the anti-VEGF burden but have not demonstrated adequate efficacy as monotherapy compared to other agents (4).
The fluocinolone acetonide intravitreal implant is being investigated in patients with both dry and exudative AMD. It is involved in a randomized, double-masked, fellow eye controlled (sham-injected) phase II clinical trial designed to evaluate changes in geographic atrophy (GA) over 24 months in patients with bilateral GA (30). It is also involved in a phase II, randomized study investigating two doses of fluocinolone acetonide and its safety and efficacy over 36 months as an adjuvant agent in patients undergoing treatment with intravitreal ranibizumab for exudative AMD (31).
Immunomodulators
Methotrexate is a dihydrofolate reductase inhibitor and inhibits T-lymphocyte proliferation by reducing nucleotide formation (4). It inhibits cytokine production and decreases inflammation. In one report, two patients with exudative AMD who were unresponsive to anti-VEGF therapy were treated with intravitreal methotrexate (32). One patient had an improved VA and both patients had decreased subretinal fluid and perifoveal leakage 2 weeks after injection.
Infliximab is a chimeric (mouse/human) IgG1 monoclonal antibody to TNF-α. It neutralizes circulating TNF-α and prevents binding to its receptors, leading to cytokine and cellular inhibition (4). TNF-α is overexpressed in CNV (17), and TNF-α inhibitors significantly reduced laser-induced CNV after intravitreal injection in animal models (33). In three patients who were treatment naive or PDT resistant, intravenous infliximab for rheumatoid arthritis was noted to improve VA and cause CNV regression after several months (34). A pilot study of systemic immunosuppression for exudative AMD found no change in the frequency of anti-VEGF injections in patients receiving intravenous infliximab (35). Intravitreal infliximab has been studied in anti-VEGF nonresponders. In one study, three patients were given intravitreal infliximab, two of whom showed complete resolution of intraretinal fluid and the third had a temporary reduction of intraretinal fluid (36). Another four patients were treated with intravitreal infliximab, without a
significant visual or anatomical benefit, but two developed severe intraocular inflammation (37).
significant visual or anatomical benefit, but two developed severe intraocular inflammation (37).
Adalimumab is another antibody to TNF-α being investigated for AMD. It is fully humanized and less likely to cause human antichimeric antibodies than is infliximab. It is undergoing phase II studies to investigate electroretinogram (ERG) abnormalities, retinal thickness, VA, and adverse events after intravitreal administration for exudative AMD (38).
Daclizumab is a fully humanized IgG1 monoclonal antibody against the α chain of the IL-2 receptor (4). This results in decreased activation and proliferation of T lymphocytes and decreased IFN-γ expression in CD4 and CD8 cells. In one trial of systemic daclizumab for exudative AMD, there was a decrease in anti-VEGF injections from 0.73 to 0.42 per month after treatment with daclizumab for 6 months (35). The authors concluded that systemic immunosuppression may alter the course of exudative AMD but that further studies are needed.
MAMMALIAN TARGET OF RAPAMYCIN
Multiple molecular pathways exist that contribute to aging, including the mammalian target of rapamycin (mTOR) pathway. The mTOR protein is a serine/threonine protein kinase that processes signals and controls cell growth and proliferation (39). Reduced mTOR activity, through knocking out downstream protein signaling or with pathway inhibitors, has been observed in mice with extended life (40). The mTOR pathway has been implicated in different cancer types and resistance to chemotherapy (41).
As the RPE ages, mitochondrial function decreases and cells are more prone to oxidative damage (39). These early changes correlate with RPE hypertrophy and dedifferentiation and coincide with mTOR activation (42). The RPE cells contain mTOR complexes that are affected by various stimuli, and down-regulation of mTOR signaling may prolong cellular survival (39). As nutrient and growth factor signals affect the mTOR pathway, it may link environmental factors to cellular functions. mTOR inhibitors down-regulate markers of cellular senescence and may have a therapeutic effect in AMD, as they may slow down RPE aging.
The mTOR pathway also has a role in angiogenesis and is affected by inflammation and complement. IFN-γ plays an important role in intraocular inflammation, and promotes VEGF secretion from RPE cells via the mTOR pathway (10). Other angiogenic factors, including TNF-α, IL-1β, IL-6, IL-8, basic fibroblast growth factor (bFGF), and VEGF, depend on the mTOR pathway for cellular signaling (43).
mTOR Inhibitors
Sirolimus (rapamycin) is a macrolide antibiotic that inhibits T-lymphocyte activation and proliferation (44). It inhibits the mTOR pathway and has antiangiogenic properties. As it has dual effects against angiogenesis and inflammation, it may be a therapeutic option for both exudative and dry AMD (10). Animal models of retinal and choroidal angiogenesis have been inhibited by sirolimus (44). It was effective in reducing VEGF from cultured RPE cells and was more effective in reducing endothelial cell sprouting than were VEGF inhibitors (45). In phase I studies in patients with diabetic macular edema, it was well tolerated and safe when administered intravitreally and subconjunctivally (46). It is being investigated in two phase I/II trials in patients with bilateral GA due to AMD. Patients will receive subconjunctival (47) or intravitreal (48) sirolimus in one eye every 2 months for 2 years, with the other eye as a control. Primary outcomes include change of GA and development of exudative AMD.
Palomid 529 is a small molecule drug that inhibits the mTOR pathway via dissociation of both target of rapamycin complexes, TORC1 and TORC2 (49). It inhibits tumor growth, angiogenesis, and vascular permeability in mutant glioma tumor cells and endothelial cells (50). As the mTOR pathway has several positive and negative feedback loops, dual TORC inhibitors may have a more effective and broad inhibition of angiogenesis than sirolimus. Phase I studies are under way to investigate safety and tolerability in patients with exudative AMD after intravitreal or subconjunctival administration (51).
COMPLEMENT
The complement system is part of the innate immune system but also has a role in adaptive immunity (5). It eliminates damaged, necrotic and apoptotic cells and facilitates the elimination of circulating immune complexes (52). This system is made of proteins that circulate normally and are activated by several triggers. Three pathways exist, and the final result is the generation of the membrane attack complex (MAC), a cell-killing protein.
The complement system is a highly regulated system but has the ability to damage host tissue (5). It may play a role in autoimmune disorders like asthma, systemic lupus erythematosus, multiple sclerosis (MS), inflammatory bowel disease, transplant rejection, and others (52). Complement-associated membranoproliferative glomerulonephritis type II is associated with deposits that resemble drusen. Complement is also thought to play a role in neurodegenerative disorders like Alzheimer’s disease (AD). Deficiencies in complement factors predispose to infections and autoimmune diseases, and complement inhibitors may cause an increased risk of infection.
Complement Pathways
The complement system is shown in Figure 25.1, including the medications under investigation and their sites of action. The classical pathway requires an antigen-antibody complex and is the only pathway that is specific for a given antigen (5). It involves the components C1, C2, and C4 and is inhibited by C1 inhibitor (C1-INH). The alternative pathway is triggered
by binding to a host cell or pathogen surface. C3b and the activated form of complement factor B (Bb) form the C3 convertase, and complement factor D is required. There is a spontaneous low-level activation of this pathway, and components of cigarette smoke can activate this pathway (53). CFH inhibits this pathway, and this is the main pathway thought to be involved in AMD (5). The lectin pathway is triggered by polysaccharides on microbial surfaces and requires C2 and C4.
by binding to a host cell or pathogen surface. C3b and the activated form of complement factor B (Bb) form the C3 convertase, and complement factor D is required. There is a spontaneous low-level activation of this pathway, and components of cigarette smoke can activate this pathway (53). CFH inhibits this pathway, and this is the main pathway thought to be involved in AMD (5). The lectin pathway is triggered by polysaccharides on microbial surfaces and requires C2 and C4.
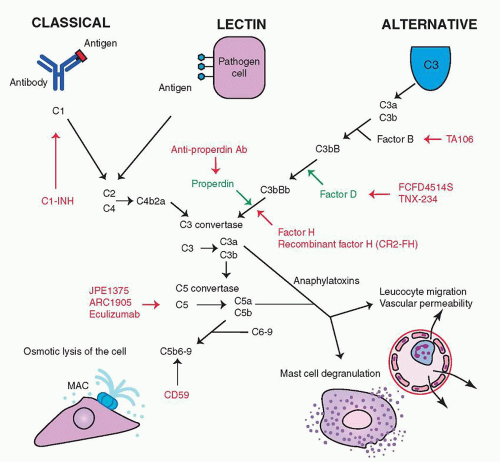 Figure 25.1 ▪ A simplified schematic of the complement cascade. The sites of action of therapies being investigated are included. |
All pathways form a C3 convertase, an enzyme that cleaves and activates C3 into C3a and C3b. C3b activates the C5 convertase, which cleaves C5 into C5a and C5b. C5b combines with C6-9, forming the terminal MAC, which causes lysis of cellular membranes and is deadly to cells and pathogens. C3a and C5a are anaphylatoxins that increase vascular permeability, cause degranulation of mast cells and neutrophils, induce cytokine release, and mediate leukocyte chemotaxis (54).
Complement and AMD
Several studies have shown that complement components may be deposited in drusen, including C3, C3a, C5a (55), and C5, the MAC (56), CFH, and complement receptor 1 (57,58). C1q and C3 have been shown to occur in CNV (9), and C3a and C5a increase VEGF production (55).
Genetic polymorphisms in complement encoding genes are associated with a predisposition for AMD. Complement factor B (CFB) and complement factor 2 genes have been associated with AMD, and genetic variations may be protective (59), as the production of the classical and alternative C3 convertases are diminished (60). Variations in the C3 gene are associated with AMD (61), as is the CFH gene; the most frequently reported factor implicated (62).
Complement Component 3
C3 is important for all three pathways, and animal and in vitro studies have demonstrated its importance in AMD. Subretinal injections of C3 in mice develop signs of AMD (63), while C3 knockout mice are protected from laserinduced CNV (64). C3a leads to increased VEGF production by RPE cells in vitro (55), and C3 has been demonstrated in surgically removed CNV (9). Plasma C3 levels appear to be elevated in patients with AMD (65).
POT-4/AL-78898A is a derivative of compstatin, a peptide inhibitor of C3 activation. Compstatin has been
shown to decrease drusen formation in monkeys with early-onset macular degeneration (66). In phase I studies, POT-4 was demonstrated to be safe when injected intravitreally (67). AL-78898A is currently in a phase II randomized, double-blinded, crossover study in patients with exudative AMD designed to demonstrate a biologic effect after one intravitreal injection (68). Outcomes include time to escape to standard of care therapy, change in retinal thickness and VA.
shown to decrease drusen formation in monkeys with early-onset macular degeneration (66). In phase I studies, POT-4 was demonstrated to be safe when injected intravitreally (67). AL-78898A is currently in a phase II randomized, double-blinded, crossover study in patients with exudative AMD designed to demonstrate a biologic effect after one intravitreal injection (68). Outcomes include time to escape to standard of care therapy, change in retinal thickness and VA.
Complement Component 5
Complement factor 5 is required for formation of the MAC. In humans, C5 is found in drusen (55), and serum levels of C5a are elevated in patients with AMD (69). C5a may increase RPE-mediated VEGF secretion (55). Mouse knockouts for the C5a receptor display reduced VEGF production and CNV formation in laser-induced models of CNV. There have not been clear associations between the C5 gene polymorphisms and AMD.
Eculizumab is a humanized monoclonal antibody against C5. It is given intravenously for paroxysmal nocturnal hemoglobinuria, another complement-mediated disorder (70).
Chronic and systemic use increases the risk of Neisseria meningitides infection. Eculizumab is being tested in a phase II, randomized, placebo-controlled, double-masked study for dry AMD (71). After receiving a meningococcal vaccine, patients are randomized to eculizumab or placebo for 24 weeks. Patients are followed for 12 months after the initial infusion, and outcome measures include changes in the size of GA, drusen volume, and VA.
ARC1905 is an anti-C5 aptamer that blocks the cleavage of C5 into C5a and C5b, thus blocking further complement activation (52). Aptamers are nonimmunogenic, singlestranded oligonucleotides that display three-dimensional folding characteristics that allow inhibition of protein-protein interactions (72). A phase I study evaluating intravitreal ARC1905 in combination with intravitreal ranibizumab for exudative AMD has been completed (73). Results have not been released. Another phase I trial is ongoing to investigate safety and tolerability of intravitreal ARC1905 in the treatment of dry AMD with drusen and/or GA (74).
JPE1375 is a small molecule peptidomimetic antagonist against the C5 receptor. This compound exhibits high stability and receptor specificity. In mice, it has been demonstrated to have activity in an immune complex-mediated peritonitis model (75).
Complement Factor D
CFD is required for the activation of the alternative pathway C3 convertase. CFD knockout mice showed a significant reduction in photoreceptor damage after being exposed to constant light for several days (76). Genetic alterations in the CFD gene may (77) or may not be associated with AMD (78). There are conflicting data on whether or not systemic CFD is elevated in AMD patients (5,69,77). Nonetheless, there still may be a therapeutic role for lowering CFD and inhibiting the alternative pathway C3 convertase.
FCFD4514S is a monoclonal antibody against CFD. A phase I study assessing the safety and tolerability, dosing, and pharmacokinetics of intravitreally delivered FCFD4514S is ongoing (79). A phase II, randomized, sham injection controlled study of safety, tolerability, and efficacy of intravitreal FCFD4514S in patients with GA is also ongoing (80). The main outcome measure is growth of GA at 12 months.
TNX-234 is a complement inhibitor that may be a therapeutic option for AMD. It is a humanized antibody against CFD (81).
Complement Factor B
CFB is a component of the alternative pathway C3 convertase (5). CFB has been localized in the neurosensory retina, in the RPE, in the choroid, and in drusen (59). In mice, its expression increases with age, and this was associated with increased C3 in the RPE and Bruch’s membrane (82). Variations in the CFB gene are associated with AMD (59) and confer a protective effect through the reduction of alternative pathway C3 convertase. In vitro studies have shown a fourfold reduction in C3b binding affinity in certain variants, and this was associated with a reduction in C3 convertase formation (60).
Complement Factor H
CFH regulates the alternative pathway by promoting the breakdown of its C3 convertase. In vivo studies have shown that CFH is produced in the RPE and accumulates in drusen, RPE cells, choroid, and extracellular space (58). CFH knockout mice demonstrate increased autofluorescent material and C3 deposition in the retina (84).
Subretinal injection of RNA directed against CFH showed increased MAC deposition and laser-induced CNV in mice (85). Genetic studies have revealed that polymorphisms in the CFH gene increase the development and progression of AMD. A substitution of tyrosine by histidine at the 402 position (Y402H) is the most reported polymorphism (62), but other variants also exist (86). High-risk CFH polymorphisms are additive with smoking (87) and may be associated with systemic complement activation (88).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


